
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Rajarshi Shahu College of Pharmacy Buldhana, Maharashtra, India
Artificial Intelligence (AI) has emerged as a transformative force in the field of drug discovery and protein design, offering innovative solutions to longstanding challenges in biomedical research. Traditional drug discovery approaches, reliant on trial-and-error, high-throughput screening, and structural biology, are often time-consuming, costly, and associated with high failure rates. AI-driven technologies, particularly machine learning (ML), deep learning (DL), and natural language processing (NLP), enable the rapid analysis of large-scale biological datasets, prediction of drug-target interactions, and design of novel compounds with optimized therapeutic potential. In drug discovery, AI contributes to target identification, virtual compound screening, lead optimization, and clinical trial design by predicting pharmacokinetics, toxicity, and efficacy at early stages. Similarly, in protein design, tools such as AlphaFold and Rosetta have revolutionized structure prediction and facilitated de novo design of functional proteins, enabling the development of novel enzymes, antibodies, and therapeutic proteins. These advances accelerate precision medicine by tailoring drugs to individual genetic and proteomic profiles while reducing development timelines and costs. Despite these breakthroughs, significant challenges remain, including the need for high-quality annotated datasets, computational limitations, biological complexity, and regulatory concerns surrounding AI-generated drug candidates. Addressing these barriers will be critical to harnessing AI?s full potential in biopharmaceutical innovation. Overall, AI-driven drug discovery and protein design represent a paradigm shift, promising faster, cost-effective, and more precise therapeutic development with profound implications for global healthcare.
Artificial Intelligence (AI) has revolutionized various fields, with biotechnology being one of the most promising. AI refers to the ability of machines to perform tasks that typically require human intelligence, such as learning, reasoning, and problem-solving. In biotechnology, AI is employed to analyze complex biological data, predict molecular behavior, and simulate biological processes, facilitating the development of new drugs and therapeutic strategies. Key AI methods used in drug discovery and protein design include machine learning (ML), deep learning (DL), and natural language processing (NLP). These methods help predict the binding of drugs to targets, optimize protein folding, and design novel compounds for therapeutic use. AI’s integration into biotechnology is not only improving efficiency but also providing new insights that were previously difficult to obtain using traditional methods (1). Historically, drug discovery and protein design were largely dependent on trial and error, high-throughput screening, and structural biology techniques such as X-ray crystallography. These methods, while groundbreaking, were time-consuming, expensive, and often resulted in limited success. The advent of AI-driven techniques marked a paradigm shift, allowing researchers to process large datasets, simulate interactions, and optimize compounds far more quickly and accurately. In the late 20th and early 21st centuries, the rise of computational biology and the increasing availability of genomic data provided the foundation for AI methods to be applied in drug discovery. Machine learning algorithms became crucial in analyzing vast amounts of genomic, proteomic, and pharmacological data to predict how drugs interact with biological targets. In protein design, AI has been instrumental in folding prediction models, such as DeepMind's AlphaFold, which has revolutionized our understanding of protein structures (2). AI’s influence in biotechnology is profound, particularly in the fields of drug discovery and protein design. Traditional drug discovery can take 10–15 years and cost billions of dollars, with no guarantee of success. AI has significantly reduced these barriers by accelerating the identification of drug candidates, optimizing their efficacy, and improving the drug development process (3). For example, AI-based models can predict the pharmacokinetics and toxicity of a drug candidate much earlier in the development cycle, allowing researchers to prioritize viable candidates. Moreover, AI’s ability to design novel proteins with desired characteristics (e.g., enzyme activity, binding affinity) has opened up new avenues for therapeutic design, especially in areas like gene editing and personalized medicine. The integration of AI into medical research not only expedites the development of new drugs but also increases the success rate of drug discovery by identifying more precise molecular targets. This results in more efficient treatments with fewer side effects and faster patient access to life-saving therapies (4). The complexity of drug discovery and protein design presents significant challenges that require innovative solutions. Traditional drug discovery processes are time-consuming, expensive, and often have high failure rates. The journey from target identification to drug approval typically spans over 10–15 years and costs billions of dollars. Moreover, the failure rate of drug candidates in clinical trials is remarkably high, often due to issues such as poor bioavailability, toxicity, or lack of efficacy. For protein design, accurately predicting protein structures and their interactions with other molecules has been a significant challenge, despite advancements in structural biology and crystallography. These complexities are exacerbated by the sheer volume of data involved in both drug discovery and protein design. The vast amount of molecular, biological, and chemical data generated during research makes it difficult for traditional methods to process and interpret efficiently. As a result, there is a growing need for more advanced technologies that can handle these large datasets and offer insights that were previously beyond reach. Drug discovery, in its traditional form, relies on a series of labor-intensive steps including target identification, compound screening, lead optimization, and clinical testing. High-throughput screening (HTS) of compounds is often used to identify potential candidates, but this process is slow and costly. Moreover, the validation of drug targets through traditional in vitro and in vivo studies is not only resource-intensive but also prone to error. Even after selecting promising candidates, the subsequent phases of drug development are fraught with uncertainties and high attrition rates. Studies show that approximately 90% of drugs fail in clinical trials, often due to issues such as inadequate efficacy or unforeseen toxicological effects [5]. In protein design, the task of predicting how a protein will fold into its three-dimensional structure is inherently difficult. Despite breakthroughs in structural biology, methods like X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy require substantial time, expense, and expertise. Furthermore, the design of novel proteins with specific functions is another area where traditional methods have limitations. These challenges have prompted researchers to seek more efficient, scalable, and cost-effective approaches to drug discovery and protein design.AI technologies, particularly machine learning (ML) and deep learning (DL), have emerged as transformative tools in both drug discovery and protein design. AI excels at processing vast datasets quickly and efficiently, identifying patterns that might be overlooked by traditional methods. By using large-scale biological data, AI models can predict the interactions between small molecules and biological targets with high precision, significantly reducing the time needed for drug development. In drug discovery, AI can be used to predict pharmacokinetics, toxicity, and efficacy of drug candidates early in the process. For instance, machine learning algorithms have been developed to predict how well a drug will bind to its target, or how it will be metabolized in the body. This allows for early-stage optimization of lead compounds, reducing the need for costly and time-consuming experimental trials. Furthermore, AI can facilitate the identification of novel drug targets by analyzing omics data, enabling the development of personalized medicine approaches that are tailored to the genetic profile of individual patients [6].In protein design, AI tools like AlphaFold have dramatically improved the accuracy of protein structure prediction. By using deep learning, AlphaFold has been able to predict protein structures from amino acid sequences with unprecedented accuracy, offering a new avenue for drug discovery. AI is also playing a crucial role in the de novo design of proteins with specific functions, such as enzymes that can catalyze reactions for industrial or therapeutic purposes [7]. These capabilities have made protein design faster, cheaper, and more scalable, holding the potential to unlock novel therapeutic approaches for diseases that were previously difficult to target. Thus, AI’s ability to enhance the speed, precision, and cost-effectiveness of drug discovery and protein design makes it an indispensable tool in modern biotechnology.
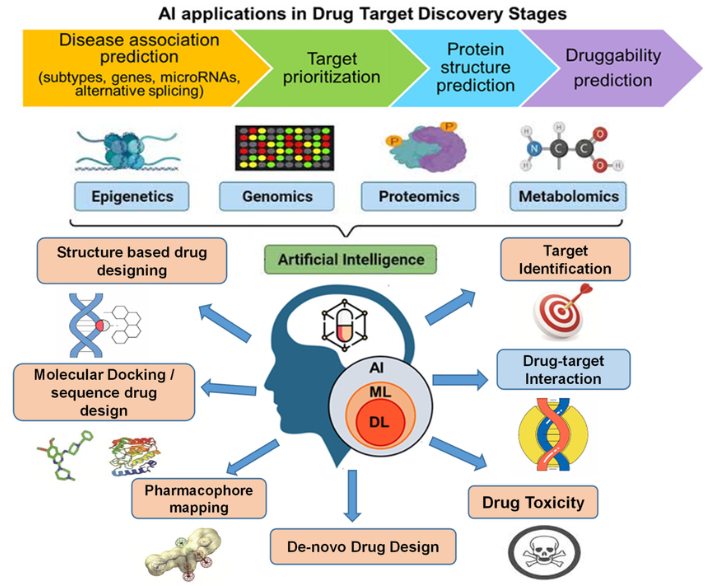
Figure 1. Artificial intelligence (AI) applications in drug target discovery stages.
2. AI IN DRUG DISCOVERY
2.1 Key Stages of Drug Discovery
The drug discovery process is a complex and multi-step journey, consisting of several critical stages. AI has been integrated into these stages to enhance the speed, precision, and efficiency of drug development. Below is an outline of each key stage and how AI is reshaping these processes.
2.1.1 Target Identification and Validation
The first stage in drug discovery is identifying the biological target that a drug can interact with to produce a therapeutic effect. Traditional methods of target identification, including genetic screenings and biochemical assays, can be time-consuming and prone to false positives. AI offers a transformative approach to this stage, allowing researchers to analyze large datasets from genomics, proteomics, and transcriptomics to pinpoint potential targets more efficiently. Machine learning models can predict the relevance of specific genes or proteins in disease pathways, suggesting novel targets for drug development. For example, deep learning techniques have been used to analyze molecular interactions and predict protein-protein interactions, enabling the identification of previously unknown therapeutic targets [8]. In addition, AI can also be employed in the validation of targets by predicting their druggability and assessing whether they can be effectively modulated by small molecules.
2.1.2 Compound Screening
Once a target is identified and validated, the next step in drug discovery is to identify small molecules that can interact with the target. Traditional high-throughput screening (HTS) involves testing thousands or even millions of compounds to find promising drug candidates, but this is a costly and time-consuming process. AI, particularly machine learning algorithms, has greatly improved the efficiency of compound screening.AI models can be trained on existing databases of chemical compounds and their biological activities to predict how new compounds might interact with the identified target. This computational approach, known as virtual screening, drastically reduces the need for wet-lab experiments and accelerates the process of finding potential drug candidates. By leveraging AI, researchers can predict the binding affinity of compounds and optimize the selection of candidates for further testing [9]. Additionally, AI models are capable of predicting the ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties of compounds early in the drug discovery process, improving the chances of identifying viable candidates.
2.1.3 Lead Optimization
Lead optimization is the stage where initial drug candidates are refined to improve their efficacy, specificity, and safety profiles. AI plays a critical role in this phase by automating the iterative process of optimizing chemical structures. Deep learning models, particularly generative models, can propose novel molecular structures that are predicted to have improved properties, such as higher binding affinity and lower toxicity.AI-driven techniques like quantitative structure-activity relationship (QSAR) modeling can analyze the relationship between a compound’s chemical structure and its biological activity. This allows researchers to optimize lead compounds based on specific biological outcomes. Furthermore, AI can be used to predict the pharmacokinetics and pharmacodynamics of drug candidates, ensuring that they will function effectively in the human body. By using these advanced AI techniques, researchers can accelerate the optimization of drug candidates, reducing the time spent on preclinical development [10].
2.1.4 Preclinical and Clinical Trials
Preclinical testing involves evaluating the safety and efficacy of lead compounds in laboratory settings (in vitro) and animal models (in vivo), while clinical trials involve testing the drug in human subjects. AI is increasingly being used to streamline both preclinical and clinical trial phases.In preclinical testing, AI can assist in the design of experiments and predict the likely outcomes based on previous data. Machine learning algorithms can analyze data from animal models and predict how the drug will behave in humans, potentially reducing the need for extensive animal testing. Additionally, AI can be used to identify biomarkers for monitoring drug efficacy and safety, ensuring that preclinical studies are more predictive of clinical success. In clinical trials, AI tools are helping with patient recruitment by identifying individuals who are most likely to respond to a treatment based on their genetic and clinical profiles. AI-driven predictive models can also analyze clinical data in real-time to monitor patient outcomes, optimize dosing regimens, and identify adverse events more quickly. Moreover, AI has the potential to enhance the precision of clinical trials by enabling more personalized treatment strategies, allowing for better targeting of specific patient populations [11].
2.2 AI Techniques in Drug Discovery
AI techniques have profoundly impacted various stages of the drug discovery process, enhancing the speed, accuracy, and efficiency of discovering new therapeutics. Several AI methods, including machine learning (ML), deep learning (DL), and natural language processing (NLP), are widely used for data mining, target identification, molecular docking, high-throughput screening, and property optimization.
2.2.1 Data Mining & Drug Repurposing
2.2.1.1 AI for Identifying Existing Drugs for New Therapeutic Uses
Data mining, powered by AI, is an essential tool for drug repurposing—the process of identifying new uses for existing drugs. AI can analyze vast amounts of public and proprietary data, including scientific literature, clinical trial results, and molecular databases, to find connections between existing drugs and new disease indications. By employing advanced algorithms, AI can identify hidden patterns in complex datasets that human researchers might overlook. For example, AI-driven platforms have been used to identify FDA-approved drugs that could be repurposed for COVID-19, by screening databases of drugs with known mechanisms of action. The approach not only shortens the drug development timeline but also reduces the cost by leveraging existing safety and efficacy data [12]. The AI models can evaluate the drug-target interactions, biological pathways, and clinical outcomes, making repurposing a faster, more efficient alternative to traditional drug discovery.
2.2.1.2 Applications in Identifying Novel Drug Candidates
Beyond repurposing, AI is also instrumental in identifying novel drug candidates by uncovering new chemical entities and predicting their therapeutic potential. Machine learning models can be trained on existing data to predict which molecular structures are likely to exhibit biological activity against specific disease targets. This approach allows for virtual screening of millions of compounds, vastly accelerating the early stages of drug discovery. In practice, AI can optimize chemical libraries to suggest molecules with higher chances of success in clinical development. For example, generative models like deep learning-based neural networks are employed to design new molecules with desirable properties, such as high binding affinity, stability, and low toxicity [13].
2.2.2 Target Identification & Validation
2.2.2.1 How AI Models Identify Potential Drug Targets (Proteins, Genes)
AI models play a pivotal role in identifying new drug targets, particularly proteins and genes implicated in disease pathways. Traditional methods of target identification rely heavily on experimental biology, which can be time-consuming and expensive. AI accelerates this process by analyzing large datasets, including genomics, proteomics, and transcriptomics, to identify genes or proteins associated with disease. Machine learning techniques, such as clustering and classification algorithms, help researchers identify disease-relevant targets by analyzing gene expression profiles and protein interactions. By leveraging these computational tools, AI models can predict which targets are most likely to lead to successful therapeutic interventions. For instance, in cancer research, AI models have been used to identify specific mutations and gene fusions in cancer cells that could be targeted by new drugs [14].
2.2.2.2 Examples of Successful Applications (e.g., Targeting Cancer, COVID-19)
AI has been successfully applied to target identification in several therapeutic areas. One prominent example is the application of AI to target novel cancer pathways, where AI tools have been used to predict biomarkers for specific types of cancer. For instance, AI models have been used to identify biomarkers in melanoma and breast cancer, paving the way for the development of targeted therapies [15]. Similarly, AI models were instrumental in rapidly identifying therapeutic targets for COVID-19. Researchers used AI to identify viral proteins as potential drug targets and to repurpose existing drugs for the treatment of COVID-19, significantly shortening the timeline for drug discovery in the face of the pandemic [16].
2.2.3 High-Throughput Screening (HTS)
2.2.3.1 AI’s Role in Automating and Enhancing HTS Techniques
High-throughput screening (HTS) is a critical method for testing large libraries of compounds to identify potential drug candidates. Traditionally, HTS is labor-intensive and expensive. AI significantly enhances this process by automating data analysis, enabling faster and more accurate identification of promising compounds. AI algorithms can process vast amounts of screening data, identifying the most promising compounds based on their ability to interact with the target protein. This reduces the need for manual analysis and speeds up the screening process. Additionally, AI-based image recognition can improve the detection of biological activity in assay plates, increasing the throughput and precision of HTS methods [17].
2.2.3.2 Predictive Modeling for Compound Efficacy
AI also contributes to HTS through predictive modeling, which estimates the efficacy of compounds based on their chemical structure and interaction with biological targets. Deep learning models can predict how compounds will behave in the human body, offering insights into their potential effectiveness, bioavailability, and toxicity. This predictive approach allows researchers to focus on compounds with the highest likelihood of success, reducing the need for expensive and time-consuming experimental trials.
2.2.4 Molecular Docking & Simulation
2.2.4.1 AI-Driven Molecular Docking for Drug-Receptor Interaction Predictions
Molecular docking is a critical technique in drug discovery, used to predict how a small molecule (e.g., a drug) binds to its target receptor. AI has enhanced this process by improving the accuracy of docking predictions and speeding up the simulations. By utilizing deep learning and reinforcement learning, AI algorithms can predict how small molecules interact with receptor sites, offering insights into binding affinity and specificity. For example, AI-based molecular docking has been used to discover inhibitors for enzymes like proteases in viral pathogens such as HIV and SARS-CoV-2 [18]. AI-enhanced docking studies have helped researchers design more potent inhibitors with higher specificity, improving the chances of clinical success.
2.2.4.2 Case Studies Demonstrating AI-Enhanced Molecular Simulations
Several case studies highlight the power of AI in molecular simulations. For instance, AI models have been used to design small molecule inhibitors for protein targets involved in Alzheimer's disease. By simulating the interactions between drug candidates and amyloid-beta peptides, AI-driven simulations identified lead compounds that showed promise in preclinical testing [19].
2.2.5 Drug Property Optimization
2.2.5.1 AI in Predicting Properties Like Toxicity, Bioavailability, and Solubility
Drug property optimization is a critical stage in the development of new therapeutics. AI plays a significant role in predicting essential drug properties, such as toxicity, bioavailability, solubility, and permeability. These predictions help researchers identify drug candidates with favorable pharmacokinetic profiles before they move on to expensive animal or clinical trials. Machine learning models are trained on large datasets of chemical structures and corresponding properties, enabling them to predict how a new compound will behave in the human body. This allows for more efficient optimization of drug candidates, reducing the need for extensive physical testing [20].
2.2.5.2 Reducing Physical Testing and Optimizing Drug Design
AI models also streamline the drug design process by suggesting modifications to chemical structures that optimize desired properties. By using AI-based tools, researchers can reduce the number of compounds that need to undergo physical testing, saving both time and resources. This is particularly beneficial in the early stages of drug discovery, where high failure rates can be mitigated by better-informed design decisions.
2.3 Deep Learning & Generative Models in Drug Discovery
Deep learning (DL) and generative models have become central to modern drug discovery. These models, powered by vast datasets and advanced algorithms, have revolutionized the way researchers identify drug candidates, optimize their properties, and predict their efficacy. Below, we explore how deep learning models and generative approaches are being used in the search for novel therapeutics.
2.3.1 Deep Learning Models
2.3.1.1 Convolutional Neural Networks (CNNs) and Recurrent Neural Networks (RNNs) in Molecular Biology
Deep learning models, particularly convolutional neural networks (CNNs) and recurrent neural networks (RNNs), have shown remarkable success in molecular biology. These models are especially valuable for analyzing and interpreting biological sequences, chemical structures, and large-scale datasets in drug discovery.
Convolutional Neural Networks (CNNs): Originally developed for image processing, CNNs have been adapted to handle molecular data by interpreting molecular structures as images or graphs. For example, CNNs are used to predict molecular properties, such as toxicity, solubility, and binding affinity, by analyzing 2D and 3D representations of molecules. The ability of CNNs to automatically learn hierarchical features has made them particularly useful in predicting complex interactions between drug molecules and their targets [21]. In some applications, CNNs can identify pharmacophores (the parts of the molecule responsible for biological activity) and optimize lead compounds for better binding and efficacy.
Recurrent Neural Networks (RNNs): RNNs are designed to process sequential data, making them ideal for tasks like predicting protein sequences or analyzing chemical reactions that follow a specific order. In drug discovery, RNNs have been employed to predict how a series of amino acids in a protein will fold, which is crucial for understanding how proteins interact with drugs. They have also been used in de novo drug design, where the network learns to generate novel molecular sequences based on known bioactive compounds [22]. This sequential modeling approach has opened new pathways for predicting the biological effects of molecular interactions.
2.3.2 Generative Models & Reinforcement Learning
2.3.2.1 Use of GANs and RL in Generating Novel Drug Molecules
Generative models, particularly Generative Adversarial Networks (GANs) and Reinforcement Learning (RL), have been applied to generate novel drug candidates and optimize drug design processes.
Generative Adversarial Networks (GANs): GANs consist of two networks— a generator that creates new data (molecules) and a discriminator that evaluates how realistic the generated data is. In drug discovery, GANs can generate entirely new molecular structures by learning the statistical properties of known compounds. This method allows for the design of molecules with specific properties, such as high binding affinity to a particular protein target or low toxicity. GANs have been used to propose novel drug candidates for diseases like cancer, HIV, and Alzheimer's, where traditional drug discovery methods may fall short [23]. By training GANs on large chemical datasets, researchers have been able to rapidly generate compounds that meet specific criteria, reducing the time needed for early-stage drug discovery.
Reinforcement Learning (RL): In the context of drug discovery, RL involves training an AI agent to optimize a drug molecule's properties through iterative trials. The agent explores the chemical space and learns to refine the molecular structure based on feedback from the environment (e.g., biological assays or predicted binding affinities). This approach can be used to generate molecules with improved efficacy, reduced toxicity, and better pharmacokinetic properties. RL is particularly effective in lead optimization, where the goal is to improve the properties of an initial molecule while maintaining its activity against the target. RL techniques have been successfully used in designing protein inhibitors and optimizing small molecule drugs [24].
2.3.2.2 Key Examples of AI-Designed Molecules Showing Promise in Clinical Trials
AI-generated molecules have made significant strides toward clinical applications, with several promising compounds emerging from AI-driven drug discovery efforts.
Baricitinib (Olumiant): One notable example of AI-assisted drug development reaching clinical success is Baricitinib, an AI-designed drug for rheumatoid arthritis. Baricitinib was designed using a machine learning algorithm to predict molecules with favorable pharmacokinetics and high target specificity. The drug was later approved by the FDA and has shown promise in treating other conditions, such as COVID-19 [25].
Insilico Medicine’s AI-Designed Molecule: Insilico Medicine, a leading company in AI-driven drug discovery, has used deep learning models to design a novel drug candidate targeting a specific protein involved in fibrosis. Their AI-designed molecule entered preclinical testing in a remarkably short time frame and demonstrated promising results in inhibiting disease progression [26]. This example highlights the power of AI in accelerating the development of highly specific therapeutics.
AI-Designed Cancer Drug Candidates: Generative models have also been employed to design small molecules targeting cancer pathways. For example, researchers have used AI to design molecules that inhibit cancer-specific protein-protein interactions. Early-stage clinical trials of these AI-generated molecules have shown encouraging results, particularly in the context of targeting hard-to-drug cancer proteins like KRAS [27].
3. AI IN PROTEIN DESIGN
3.1 Importance of Protein Design in Drug Development
Proteins are central to almost all biological processes and play a pivotal role in the mechanisms of disease. The development of therapeutic agents that target specific proteins has been a cornerstone of drug discovery. Proteins serve as both targets and effectors in the treatment of many diseases, making protein design an essential aspect of developing new and effective drugs.
3.1.1 Role of Proteins in Disease Mechanisms and Therapeutic Strategies
Proteins are involved in nearly every aspect of cell function, from catalyzing biochemical reactions as enzymes to transmitting signals as receptors, and regulating gene expression as transcription factors. Many diseases, including cancers, neurodegenerative disorders, and infectious diseases, arise due to abnormalities in protein functions. These abnormalities can include genetic mutations that alter protein structure, function, or interactions, making them potential drug targets.
Proteins as Therapeutic Targets: In cancer, for instance, abnormal proteins such as mutant p53 or altered kinase activity (e.g., BCR-ABL in chronic myeloid leukemia) contribute to the uncontrolled cell growth and proliferation seen in tumors. Designing drugs that can bind to and inhibit the abnormal function of these proteins is critical in cancer therapy [28].
Enzyme Inhibition: Many therapeutic drugs are designed to target enzymes that are overactive or mutated in diseases. For example, protease inhibitors, such as those used in the treatment of HIV and Hepatitis C, bind to viral enzymes, preventing them from processing viral proteins and inhibiting viral replication [29].
Protein-Protein Interactions: In neurodegenerative diseases like Alzheimer's, proteins such as amyloid-beta and tau form harmful aggregates that disrupt normal cellular functions. Therapeutic strategies involve designing molecules that can either stabilize the correct folding of these proteins or prevent their aggregation [30]. Similarly, protein-protein interactions (PPIs) are critical in immune responses and cancer cell metastasis, making their inhibition or modulation a promising therapeutic strategy.
3.1.1. AI’s Role in Protein-Based Drug Development
AI and machine learning (ML) are proving to be transformative tools in protein design. With the growing understanding of how proteins interact with small molecules and other biomolecules, AI can predict and optimize protein-ligand interactions, making the drug design process more efficient. AI models are employed for:
Target Identification and Validation: AI algorithms can analyze proteomic data to identify key proteins involved in disease pathways. For instance, AI models can predict new cancer biomarkers or viral proteins susceptible to inhibition [31].
Molecular Docking and Binding Affinity Prediction: AI models predict how potential drug molecules bind to a target protein. AI-based docking simulations can simulate thousands of interactions in a fraction of the time required by traditional methods, helping to identify drug candidates with high binding affinity and specificity [32].
3.1.2. Challenges in Predicting Protein Structures and Functions
One of the major challenges in protein-based drug development is the accurate prediction of protein structures and their subsequent functions. Despite significant advances in technology and AI, several difficulties persist that limit the speed and success of drug design efforts.
Challenges in Predicting Protein Structures
Proteins are composed of long chains of amino acids that fold into complex three-dimensional (3D) structures. The process of folding is highly dynamic and context-dependent, and predicting the final structure based on the linear amino acid sequence is a problem that has puzzled scientists for decades.
Protein Folding Problem: The "protein folding problem" refers to the challenge of predicting the 3D structure of a protein solely from its amino acid sequence. Although recent advances, such as DeepMind's AlphaFold, have made significant strides in predicting protein structures with remarkable accuracy, there are still many challenges. AlphaFold has demonstrated that AI can predict protein structures at a level that rivals traditional experimental methods, but these predictions are still limited by the complexity of certain protein families, especially membrane proteins and multi-subunit complexes [33].
Structural Flexibility: Another issue is the flexibility of protein structures. Proteins are not static; they often undergo conformational changes in response to environmental factors, binding with ligands, or protein-protein interactions. Predicting these dynamic states is crucial for designing drugs that can effectively modulate protein functions. AI has been increasingly applied to this task, but accurately modeling protein flexibility remains a challenge [34].
Membrane Proteins and Complex Structures: Membrane proteins, which make up about 30% of the human proteome, are notoriously difficult to study due to their hydrophobic nature. Predicting their structure and function using AI remains an area of active research. Similarly, the prediction of complex multi-subunit protein structures, such as ribosomes and proteasomes, is a difficult task for AI models due to the intricate interplay between protein subunits [35].
Challenges in Predicting Protein Functions
Even when a protein's structure is known, predicting its function is often more challenging. While structures can be inferred from sequence data, functional predictions require a deep understanding of the protein’s interactions with other molecules, its role in cellular pathways, and its behavior in a given biological context.
Functional Annotation: AI tools, such as protein sequence alignment and molecular dynamics simulations, can assist in annotating the potential functions of proteins. However, many proteins still lack functional annotation, especially those involved in complex cellular processes. This is particularly true for orphan proteins that have no known homologs or established pathways [36].
Context-Dependent Functionality: The function of a protein often depends on its cellular context and its interactions with other proteins or small molecules. This makes it challenging to predict how a protein will behave under different conditions, such as in the presence of a drug or in disease states. AI models can simulate such interactions, but comprehensive models that integrate all possible variables are still under development [37].
Post-Translational Modifications (PTMs): Post-translational modifications, such as phosphorylation, glycosylation, and ubiquitination, significantly influence protein function and stability. Predicting how these modifications affect protein activity is an area where AI can help, but current models still face limitations in accurately predicting the outcomes of PTMs on protein function and drug interactions [38].
3.2 AI Methods for Protein Structure Prediction
Understanding the three-dimensional (3D) structure of proteins is essential for deciphering their functions, predicting interactions with other biomolecules, and designing drugs that can effectively target them. Traditional methods for protein structure prediction, such as X-ray crystallography and nuclear magnetic resonance (NMR), are time-consuming and expensive. AI-driven approaches, however, have revolutionized protein structure prediction, enabling faster and more accurate predictions.
3.2.1 AlphaFold and the Revolution in Protein Folding
3.2.1.1 DeepMind’s AlphaFold: Overview, Significance, and Achievements
AlphaFold, developed by DeepMind, represents a groundbreaking achievement in the field of protein structure prediction. It employs deep learning techniques to predict the 3D structures of proteins from their amino acid sequences with remarkable accuracy.
Overview and Approach: AlphaFold uses a novel deep learning architecture that integrates data from multiple sources, including evolutionary information from protein sequences and structural templates. The model leverages the large-scale protein sequence database and protein structure data available to train on millions of sequences and known structures, allowing it to predict how proteins fold in a way that mirrors natural biological processes [39].
Significance and Achievements: AlphaFold's success was demonstrated in the 13th Critical Assessment of Structure Prediction (CASP13) competition, where it outperformed all previous methods and predicted protein structures with a level of accuracy comparable to experimental techniques. This achievement is seen as a milestone in solving the "protein folding problem," which had challenged biologists for decades. AlphaFold's predictions are particularly accurate for proteins that do not have a close homologous structure, where traditional methods have struggled [40].
Impact on Drug Discovery: The ability to predict protein structures accurately and rapidly has profound implications for drug discovery. It enables the identification of new drug targets, the design of small molecules that can interact with specific protein sites, and the optimization of drug candidates. By providing detailed insights into protein structures, AlphaFold has made it possible to accelerate the drug development process and improve the precision of drug design [41].
3.2.1.2 Impact of AlphaFold on Solving the Protein Folding Problem
AlphaFold's impact on the protein folding problem cannot be overstated. The "folding problem" refers to the challenge of predicting how a protein's linear sequence of amino acids folds into its final 3D shape, a crucial step for understanding its function.
Faster and More Accurate Predictions: AlphaFold’s deep learning-based method has drastically reduced the time required for protein structure prediction, from years of experimentation to mere hours or days of computational work. It provides a more accurate model of protein structures than earlier computational approaches, even in cases where experimental data was lacking [42].
Application to Human Proteome: One of the most important outcomes of AlphaFold’s development is its application to the entire human proteome. In 2021, DeepMind and EMBL (European Molecular Biology Laboratory) released predictions for the structure of nearly every human protein, representing a significant leap in the field of structural biology. This resource is invaluable for researchers working on drug discovery, as it helps to create a comprehensive map of the human proteome and identify potential druggable targets [43].
Protein Folding in Disease: By predicting how proteins fold and interact with each other, AlphaFold can assist in understanding how misfolded proteins contribute to diseases like Alzheimer's, Parkinson's, and cystic fibrosis. It can also aid in the development of therapies aimed at preventing or reversing protein misfolding [44].
3.2.2 Rosetta and Protein Structure Prediction
3.2.2.1 Use of AI-enhanced Rosetta for Protein Modeling and Design
Rosetta is a widely used software suite for protein structure prediction, design, and analysis. While it has been around for over two decades, recent advancements in AI and machine learning have significantly enhanced its capabilities.
AI Enhancements in Rosetta: Traditionally, Rosetta used a physics-based approach to predict protein structures by simulating the folding of polypeptides. However, recent improvements have incorporated AI algorithms to optimize the design of proteins and peptides. By leveraging machine learning, Rosetta can more efficiently explore the vast conformational space of proteins and design novel sequences with desired structural and functional properties [45].
Applications in Drug Design: AI-enhanced Rosetta is particularly useful in designing proteins that can bind to specific targets, such as in the development of antibody therapeutics or enzyme inhibitors. By modeling protein-ligand interactions, Rosetta allows researchers to design more potent and selective drug candidates. Additionally, the integration of AI enables Rosetta to design de novo protein structures with novel functions, expanding the potential for protein-based therapies [46].
Rosetta in Protein Engineering: Beyond structure prediction, Rosetta has been utilized in protein engineering to design proteins with specific stability, binding affinity, or catalytic activity. This approach is used in creating synthetic enzymes or engineered antibodies for therapeutic applications [47].
3.2.3 Protein-Protein Interaction Prediction
3.2.3.1 AI-driven Methods for Understanding Protein Interactions and Designing Inhibitors
Proteins rarely function in isolation; they often interact with other

Sakshi Tayde, Janki Wankhade, Sakshi Bendarkar, Ashwini Patil, Poonam Tale, Ashwini Verulkar, Mohini Kale, Vaishnavi Kamble, Shivshankar Nagrik*, A Comprehensive Review on Artificial Intelligence for Accelerating Drug Discovery and Protein Design, Int. J. Med. Pharm. Sci., 2025, 1 (10), 43-71. https://doi.org/10.5281/zenodo.17341812
 10.5281/zenodo.17341812
10.5281/zenodo.17341812