
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Assistant Professor, Department of Pharmaceutical Chemistry, S.R.R. College of Pharmaceutical Sciences, Valbapur, Warangal – 505476.
2Assistant Professor, Department of Pharmaceutical Chemistry, Talla Padmavathi College of Pharmacy, Warangal – 506002.
3Professor, Department of Pharmaceutical Chemistry, Telangana Social Welfare Residential Pharmacy College for Women, Anantharam, Mahabubabad – 506101.
Balofloxacin is a third-generation fluoroquinolone antibiotic widely used for the treatment of urinary tract and respiratory tract infections caused by susceptible bacterial pathogens. The development of reliable, rapid, and stability-indicating analytical methods is essential to ensure the quality, safety, and efficacy of pharmaceutical formulations during manufacturing and storage. Although several analytical methods have been reported for the estimation of balofloxacin, there remains a need for a simple, accurate, precise, and validated RP-HPLC method suitable for routine quality control analysis. Therefore, the present study was undertaken to develop and validate a stability-indicating reverse-phase high-performance liquid chromatographic (RP-HPLC) method for the quantitative estimation of balofloxacin in bulk drug and tablet dosage forms in accordance with International Council for Harmonisation (ICH) guidelines. Chromatographic separation was achieved using a Qualisil BDS C18 column (250 × 4.6 mm, 5 µm) with a mobile phase consisting of acetonitrile and phosphate buffer (75:25, v/v; pH 3.0) at a flow rate of 1.0 mL/min, and detection was carried out at 293 nm using a photodiode array detector. The developed method was validated for system suitability, specificity, linearity, accuracy, precision, robustness, ruggedness, limit of detection, and limit of quantification. The calibration curve exhibited excellent linearity over the concentration range of 2–12 µg/mL with a correlation coefficient (r²) of 0.999. Recovery studies demonstrated excellent accuracy with recoveries ranging from 99.56% to 100.09%, while precision studies showed percentage relative standard deviation values below 2%, confirming the repeatability and reliability of the method. The limits of detection and quantification were found to be 0.21 µg/mL and 0.66 µg/mL, respectively. Forced degradation studies performed under acidic, alkaline, oxidative, thermal, and photolytic stress conditions confirmed that the method effectively separated balofloxacin from its degradation products, demonstrating its stability-indicating capability. The validated RP-HPLC method was found to be simple, rapid, accurate, precise, robust, and suitable for routine quality control analysis, stability studies, and assay of balofloxacin in bulk drug and pharmaceutical dosage forms.
The pharmaceutical industry places significant emphasis on ensuring the quality, safety, and efficacy of medicinal products throughout their shelf life. Reliable analytical methods are indispensable for the identification, quantification, and quality assessment of active pharmaceutical ingredients (APIs), impurities, and degradation products during drug development, manufacturing, and post-marketing surveillance. Among the various analytical techniques available, reverse-phase high-performance liquid chromatography (RP-HPLC) has become one of the most widely employed methods owing to its excellent selectivity, sensitivity, reproducibility, and suitability for routine pharmaceutical quality control [1, 9, 16, 20, 21]. Fluoroquinolones constitute an important class of broad-spectrum antibacterial agents extensively used in the treatment of bacterial infections. Balofloxacin, a third-generation fluoroquinolone antibiotic, exhibits potent antimicrobial activity against both Gram-positive and Gram-negative microorganisms by inhibiting bacterial DNA gyrase (topoisomerase II) and topoisomerase IV, thereby preventing bacterial DNA replication and cell division. Owing to its favorable pharmacokinetic profile, broad antibacterial spectrum, and therapeutic effectiveness, balofloxacin has gained considerable clinical importance in the treatment of respiratory tract infections, urinary tract infections, skin infections, and other susceptible bacterial diseases [6, 22–24, 40]. The therapeutic efficacy of pharmaceutical products largely depends on maintaining the chemical integrity and stability of the active pharmaceutical ingredient throughout manufacturing, transportation, storage, and distribution. Exposure to environmental factors such as heat, light, moisture, acidic or alkaline conditions, and oxidative agents may result in chemical degradation, leading to reduced drug potency, altered pharmacological activity, or the formation of potentially toxic degradation products. Consequently, regulatory agencies recommend the development of stability-indicating analytical methods capable of accurately quantifying the intact drug while effectively resolving degradation products generated under various stress conditions [2–5, 18, 19, 39]. Reverse-phase high-performance liquid chromatography has become the preferred analytical technique for pharmaceutical quality control because it provides rapid analysis, high resolution, excellent precision, and superior sensitivity for the simultaneous separation and quantification of pharmaceutical compounds and their related impurities. Furthermore, RP-HPLC methods are readily adaptable for routine industrial quality control and regulatory compliance because they can be validated according to internationally accepted guidelines. The flexibility of RP-HPLC in terms of column chemistry, mobile phase composition, detector selection, and optimization parameters makes it highly suitable for the analysis of complex pharmaceutical formulations [1, 9, 17, 20, 27, 36].
Several analytical techniques, including ultraviolet (UV) spectrophotometry, high-performance thin-layer chromatography (HPTLC), liquid chromatography–mass spectrometry (LC–MS), and RP-HPLC, have been reported for the estimation of balofloxacin in pharmaceutical dosage forms and biological matrices. Although these methods have demonstrated acceptable analytical performance, several reported procedures require lengthy chromatographic run times, complex mobile phase compositions, expensive instrumentation, or extensive sample preparation. In addition, many published methods lack comprehensive forced degradation studies and complete validation according to the International Council for Harmonisation (ICH) recommendations, thereby limiting their applicability as true stability-indicating methods for routine quality control analysis [6, 7, 22–26, 33]. According to the International Council for Harmonisation (ICH), analytical procedures intended for pharmaceutical quality control should be validated for specificity, system suitability, linearity, accuracy, precision, robustness, ruggedness, limit of detection, limit of quantification, and analytical range before they can be considered suitable for regulatory applications. Furthermore, forced degradation studies under acidic, alkaline, oxidative, thermal, photolytic, and neutral stress conditions are recommended to establish the stability-indicating capability of the developed analytical method. Such validation ensures that the analytical procedure consistently produces reliable, reproducible, and scientifically acceptable results suitable for routine pharmaceutical analysis and regulatory submissions [4, 5, 18, 19, 39]. Recent advancements in chromatographic science have focused on developing analytical methods that are not only accurate and precise but also rapid, economical, environmentally sustainable, and suitable for routine laboratory applications. Modern pharmaceutical industries increasingly require analytical procedures that reduce solvent consumption, shorten analysis time, improve chromatographic resolution, and provide enhanced reproducibility without compromising analytical performance. Consequently, considerable research efforts have been directed toward optimizing chromatographic parameters, including mobile phase composition, pH, stationary phase selection, flow rate, and detection wavelength, to achieve efficient and reliable analytical methods for pharmaceutical compounds [10, 11, 20, 21, 28, 30, 34, 35]. Although several analytical procedures for balofloxacin have been published, there remains a need for a simple, rapid, economical, robust, and fully validated RP-HPLC method capable of accurately estimating balofloxacin in bulk drug and tablet dosage forms while effectively separating degradation products generated under various stress conditions. Such a method would provide a practical analytical tool for routine quality control, stability testing, and regulatory evaluation in pharmaceutical industries and research laboratories [6, 7, 22–26, 40]. Therefore, the present investigation was undertaken to develop and validate a simple, rapid, accurate, precise, economical, and stability-indicating RP-HPLC method for the quantitative estimation of balofloxacin in bulk drug and pharmaceutical tablet dosage forms. The developed method was optimized by evaluating various chromatographic parameters, including mobile phase composition, pH, flow rate, and detection wavelength, and subsequently validated in accordance with ICH guidelines. Furthermore, forced degradation studies were performed under acidic, alkaline, oxidative, thermal, photolytic, and neutral stress conditions to establish the stability-indicating capability of the proposed analytical method and demonstrate its suitability for routine pharmaceutical quality control and stability assessment [4, 5, 18, 19, 39].
MATERIALS AND METHODS
2.1 Chemicals and Reagents
Table 1. Chemicals and Reagents Used in the Study
|
Sr. No. |
Chemical/Reagent |
Grade |
Manufacturer/Supplier |
Purpose |
|
1 |
Balofloxacin Reference Standard |
Reference Standard |
Alkem Laboratories Ltd., Mumbai, India |
Standard drug |
|
2 |
Balofloxacin Tablets |
Commercial Formulation |
Procured from Local Market |
Sample analysis |
|
3 |
Acetonitrile |
HPLC Grade |
Merck Chemicals Ltd., Mumbai, India |
Mobile phase preparation |
|
4 |
Potassium Dihydrogen Phosphate |
AR Grade |
Standard Laboratory Reagent |
Buffer preparation |
|
5 |
Orthophosphoric Acid |
AR Grade |
Standard Laboratory Reagent |
pH adjustment |
|
6 |
Double-Distilled Water |
Laboratory Grade |
In-house |
Mobile phase preparation |
|
7 |
Membrane Filter (0.45 µm) |
HPLC Compatible |
Millipore/Equivalent |
Filtration of mobile phase and samples |
2.2 Instrumentation
Table 2. Instruments Used in the Study
|
Sr. No. |
Instrument |
Specification/Description |
Purpose |
|
1 |
HPLC System |
Shimadzu LC-20 HPLC System |
Chromatographic analysis |
|
2 |
Solvent Delivery System |
Quaternary Pump |
Delivery of mobile phase |
|
3 |
Detector |
Photodiode Array (PDA) Detector |
Detection of analyte |
|
4 |
HPLC Column |
Qualisil BDS C18 (250 mm × 4.6 mm i.d., 5 µm) |
Separation of balofloxacin |
|
5 |
Analytical Balance |
Sensitivity ±0.1 mg |
Accurate weighing of chemicals and samples |
|
6 |
Digital pH Meter |
Calibrated Digital pH Meter |
Adjustment of buffer pH |
|
7 |
Ultrasonic Bath |
Laboratory Ultrasonic Cleaner |
Sonication of standard and sample solutions |
|
8 |
Vacuum Filtration Assembly |
0.45 µm Membrane Filtration Unit |
Filtration of mobile phase and samples |
|
9 |
Volumetric Glassware |
Class A Volumetric Flasks, Pipettes and Measuring Cylinders |
Preparation of analytical solutions |
2.3 Chromatographic Conditions
Table 3. Optimized RP-HPLC Chromatographic Conditions for the Determination of Balofloxacin
|
Parameter |
Optimized Condition |
|
Chromatographic system |
Shimadzu LC-20 Binary Gradient HPLC |
|
Detector |
PDA Detector |
|
Column |
Qualisil BDS C18 (250 × 4.6 mm, 5 µm) |
|
Mobile phase |
Acetonitrile : 0.02 M Sodium Dihydrogen Phosphate Buffer (75:25, v/v) |
|
Buffer pH |
3.0 (adjusted with Orthophosphoric acid) |
|
Flow rate |
1.0 mL/min |
|
Detection wavelength |
293 nm |
|
Injection volume |
20 µL |
|
Run time |
10 min (or actual run time used) |
|
Retention time |
3.20 min |
|
Column temperature |
Ambient |
|
Diluent |
Methanol |
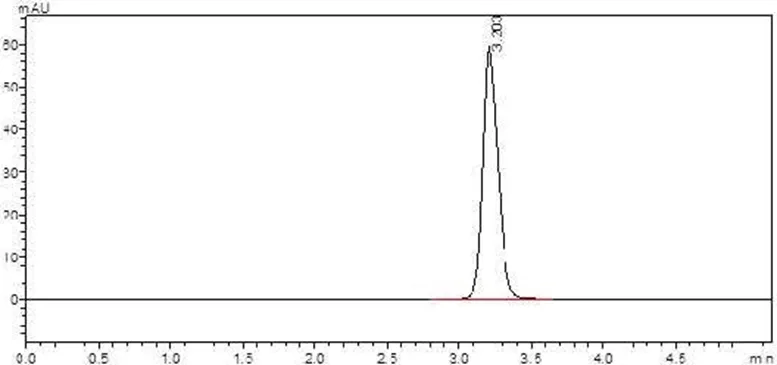
Figure 1. Representative RP-HPLC chromatogram of balofloxacin standard obtained using acetonitrile: phosphate buffer (75:25, v/v; pH 3.0) as the mobile phase, showing a retention time (tR) of 3.20 min.
2.4 Preparation of Standard Solution
An accurately weighed quantity of 25 mg of balofloxacin reference standard was transferred into a 25 mL volumetric flask. The drug was dissolved in a small volume of acetonitrile with sonication, and the volume was adjusted to the mark using the same solvent to obtain a standard stock solution containing 1000 µg/mL of balofloxacin. A suitable aliquot of the stock solution was further diluted with the mobile phase to prepare the working standard solution. Serial dilutions were prepared using the mobile phase to obtain calibration standards in the concentration range of 2–12 µg/mL. All prepared solutions were filtered through a 0.45 µm membrane filter prior to chromatographic analysis.
2.5 Preparation of Sample Solution
Twenty commercially available balofloxacin tablets were accurately weighed individually, and the average tablet weight was determined. The tablets were finely powdered using a clean mortar and pestle. An accurately weighed quantity of tablet powder equivalent to 25 mg of balofloxacin was transferred into a 25 mL volumetric flask. Approximately 15 mL of acetonitrile was added, and the mixture was sonicated for 10–15 min to ensure complete extraction of the drug. After cooling to room temperature, the volume was made up to the mark with acetonitrile to obtain the sample stock solution. The resulting solution was filtered through a 0.45 µm membrane filter, and an appropriate aliquot was further diluted with the mobile phase to obtain the required working concentration for chromatographic analysis. The prepared sample solution was injected into the RP-HPLC system, and the assay was performed by comparing the peak area of the sample with that of the corresponding standard solution.
2.6 Method Development and Optimization
The RP-HPLC method was systematically developed and optimized to obtain satisfactory chromatographic performance for the quantitative estimation of balofloxacin. Various chromatographic parameters, including the stationary phase, mobile phase composition, buffer pH, flow rate, and detection wavelength, were investigated to achieve optimum separation, acceptable retention time, symmetrical peak shape, and adequate resolution. The final chromatographic conditions were selected based on their ability to produce sharp, well-resolved peaks with excellent reproducibility and minimal analysis time, making the method suitable for routine pharmaceutical quality control.
Table 4. Parameters Evaluated During RP-HPLC Method Development and Optimization
|
Sr. No. |
Parameter Evaluated |
Purpose |
|
1 |
Selection of chromatographic column (Qualisil BDS C18, 250 × 4.6 mm, 5 µm) |
To achieve efficient separation and good peak shape |
|
2 |
Optimization of mobile phase composition |
To obtain adequate peak separation and acceptable resolution |
|
3 |
Optimization of buffer pH |
To improve peak symmetry, retention, and analyte stability |
|
4 |
Optimization of flow rate |
To obtain an appropriate retention time with satisfactory chromatographic performance |
|
5 |
Selection of detection wavelength (293 nm) |
To achieve maximum detector response and sensitivity |
|
6 |
Optimization of injection volume |
To obtain reproducible peak areas without peak distortion |
|
7 |
Evaluation of retention time |
To ensure consistent elution of balofloxacin |
|
8 |
Assessment of peak symmetry and peak shape |
To obtain symmetr |
2.7 Method Validation
The developed RP-HPLC method was validated in accordance with the International Council for Harmonisation (ICH) Q2(R2) guidelines to establish its suitability for the quantitative estimation of balofloxacin in pharmaceutical dosage forms. The validation parameters evaluated included system suitability, specificity, linearity, accuracy, precision, intermediate precision, robustness, ruggedness, limit of detection (LOD), and limit of quantification (LOQ). The validation studies demonstrated that the developed method was reliable, accurate, precise, reproducible, and suitable for routine quality control analysis.
Table 5. Validation Parameters Evaluated According to ICH Q2(R1) Guidelines
|
Validation Parameter |
Acceptance Criteria |
|
Specificity |
No interference at analyte retention time/Rf |
|
Linearity |
Correlation coefficient (R² ≥ 0.999) |
|
Accuracy |
Recovery 98–102% |
|
Precision |
%RSD ≤ 2% |
|
Repeatability |
%RSD ≤ 2% |
|
Intermediate precision |
%RSD ≤ 2% |
|
Robustness |
No significant variation |
|
Ruggedness |
Comparable results between analysts/instruments |
|
Limit of Detection (LOD) |
As calculated |
|
Limit of Quantification (LOQ) |
As calculated |
|
System suitability |
Meets predefined acceptance criteria |
2.8 Forced Degradation study
Table 6. Forced Degradation Conditions Used for Stability-Indicating Method Development
|
Stress Condition |
Reagent/Condition |
Experimental Condition |
Purpose |
|
Acid hydrolysis |
0.1 N HCl |
Reflux for specified time |
Acid degradation |
|
Alkaline hydrolysis |
0.1 N NaOH |
Reflux for specified time |
Base degradation |
|
Neutral hydrolysis |
Water |
Reflux |
Hydrolytic degradation |
|
Oxidative degradation |
Hydrogen peroxide |
3% H₂O₂ |
Oxidative degradation |
|
Thermal degradation |
Dry heat |
80–105°C |
Thermal stability |
|
Photolytic degradation |
UV/Visible light |
ICH recommended exposure |
Photostability |
RESULTS AND DISCUSSION
3.1 Selection and Optimization of the mobile Phase
Different ratios of n-butanol and methanol were tried as mobile phase was tried but, tailing of spot, less persistent spots were observed in most of the attempts. In order to overcome the problems, n-butanol: methanol: ammonia (2.5:1:1.5 v/v/v) was tried and results is good resolution, sharp and symmetrical peak with Rf value of 0.53 for BLF. Table 3.2.1 and chromatogram shown in Figure 3.2.1.
Table 7: Optimization of Mobile Phase
|
Sr. No. |
Solvent System |
Composition (v/v/v) |
Rf BLF |
|
1 |
chloroform: methanol |
2.5:2.5 |
0.87 tailing |
|
2 |
n-butanol: methanol |
4:1 |
0.65 tailing |
|
3 |
n-butanol: methanol: ammonia |
2.5:1.5:1 |
0.75 tailing |
|
4 |
n-butanol : methanol: ammonia |
2.5:1:1.5 |
0.53 |
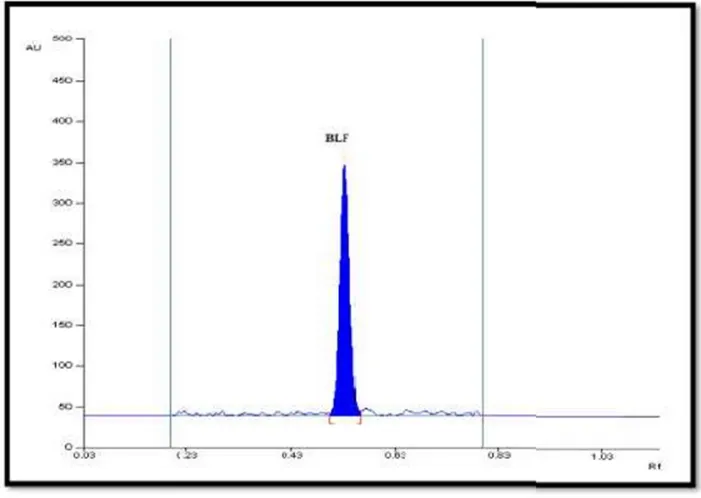
Figure3.2.1: Densitogram of standard BLF(Rf0.53±0.03), measuredat293nm, mobile phase n-butanol: methanol: ammonia (2.5: 1: 1.5 v/v/v).
3.2 Finalized chromatographic conditions
After examining the results from initial experimental conditions, the final working chromatographic conditions are summarized in Table 3.2.2.
Table 8: finalized chromatographic conditions
|
Parameters |
Specifications |
|
Stationary phase |
Aluminumbackedsilicagel60F-254 TLC plates, (10cm×10cm, layer thickness0.2mm, E-Merck, Darmstadt, Germany) prewashed with methanol |
|
Mobile phase |
n- butanol: methanol: ammonia (2.5:1:1.5v/v/v) |
|
Chamber saturation |
20 minutes |
|
Migration distance |
80 mm |
|
Activation of prewashed plate |
10 min |
|
Bandwidth |
6 mm |
|
Slit dimensions |
6.00 x0.45 mm |
|
Radiation source |
Deuterium lamp |
|
Scanning wavelength |
293 nm |
|
Distance between bands |
15.0 mm |
3.3 Linearity Study of BLF
Table 9: Linearity Study of BLF
|
Sr. No |
Concentration |
Peak area mean |
% |
|
|
in [ng/spot] |
±S.D. |
R.S.D. |
|
1 |
100 |
3928.0±66.47 |
1.16 |
|
2 |
200 |
5541.8±62.25 |
1.12 |
|
3 |
300 |
6900.6±75.92 |
1.1 |
|
4 |
400 |
8249.7±59.14 |
0.71 |
|
5 |
500 |
9596.6±83.65 |
0.87 |
|
6 |
600 |
11349±85.23 |
0.75 |
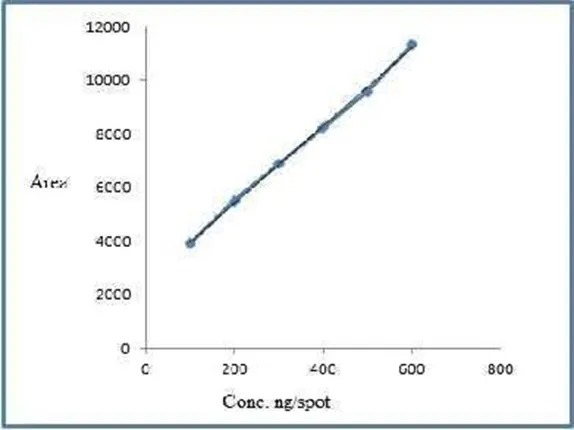
Figure3.2.2: Calibration Curve of BLF; Y=14.462X+2532.5; Where, Correlation coefficient = 0.998, Slope = 14.462, Intercept=2532.5
3.4 Analytical study
Table3.2.4: Analysis of bulk material
|
Drug |
Amount Taken (ng/band) |
Amount Found |
(ng) |
Amount Found % |
|
|
400 |
399.36 |
|
99.84 |
|
|
400 |
398.71 |
|
99.67 |
|
BLF |
400 |
399.58 |
|
99.89 |
|
|
400 |
401.44 |
|
100.36 |
|
|
400 |
399.92 |
|
99.98 |
|
|
400 |
400.95 |
|
100.23 |
|
|
Mean ±SD |
398.773.71 |
|
99.69 |
|
|
%RSD |
0.93 |
|
0.93 |
Brand Name: BALOKEM
Mfg. By: Alkem Lab. Ltd. Mumbai.
Batch No.: BKT1004HB
Average weight: 257.12mg
Table3.2.5: Analysis of Tablet Formulation
|
Drug |
Amount Taken (ng/band) |
Amount Found |
(ng) |
Amount Found % |
|
|
400 |
405.23 |
|
101.30 |
|
|
400 |
404.17 |
|
101.04 |
|
BLF |
400 |
395.52 |
|
98.88 |
|
|
400 |
408.81 |
|
102.20 |
|
|
400 |
402.16 |
|
100.54 |
|
|
400 |
404.23 |
|
101.05 |
|
|
Mean ± SD |
403.354.41 |
|
100.84 |
|
|
%RSD |
1.09 |
|
0.99 |
3.5 Accuracy
Table3.2.6: Recovery studies
|
Drug |
Initial Amount [ng/band] |
Amount added (%) |
Amount recovered ±S.D. [ng/band] [n=3] |
% Recovered |
% RSD |
|
|
200 |
80 |
362.621.29 |
100.73 |
0.35 |
|
BLF |
200 |
100 |
399.581.38 |
99.89 |
0.34 |
|
|
200 |
120 |
440.343.82 |
100.07 |
0.86 |
Table3.2.7: Precision Studies (Intra-day and Inter-day)
|
Drug |
Conc.[ng/band] |
Intra day Amount found[ng] |
Inter day Amount found[ng] |
|
|
|
Mean± SD % [n=3] RSD |
Mean± SD %RSD [n=3] |
|
|
200 |
201.47 ± 1.97 0.98 |
201.29 ± 3.10 1.54 |
|
BLF |
300 |
299.95 ± 0.97 0.32 |
300.53 ± 5.13 1.71 |
|
|
400 |
400.24 ± 4.33 1.08 |
399.45 ± 4.52 1.13 |
3.6 Repeatability Studies
Table3.2.8: Results of Repeatability Studies
|
Drug |
Amount Taken (ng/band) |
Amount Found(ng) |
Amount Found % |
|
|
400 |
401.65 |
100.41 |
|
|
400 |
400.72 |
100.18 |
|
BLF |
400 |
400.28 |
100.07 |
|
|
400 |
399.29 |
99.82 |
|
|
400 |
403.82 |
100.95 |
|
|
400 |
400.22 |
100.05 |
|
|
Mean ±SD |
401.0011.52 |
100.25 |
|
|
%RSD |
0.39 |
0.39 |
3.7 Specificity
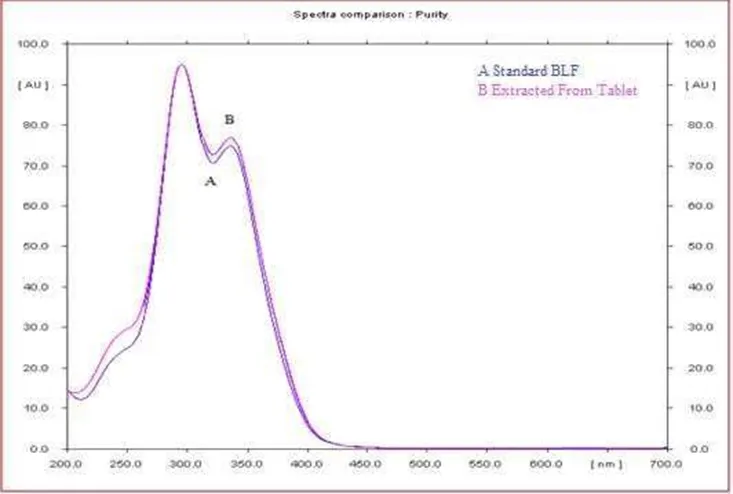
Fig 3.2.3: Peak purity spectra of standard BLF (A), sample (B) extracted from a BLF tablet, scanned at the peak-start, peak-apex, and peak-end positions of the spot (correlation > 0.99)
3.8 Summary of developed method
Table4.1: Summary of developed methods I and II
|
Parameter |
Method I |
Method II |
|
Linearity range |
2-12 µg/mL |
100-600 ng/spot |
|
Correlation coefficient |
0.999 |
0.998 |
|
Linearity[equation] |
y=96956x+101883 |
y=14.462x+2532.5 |
|
LOD |
0.21 µg |
8.99 ng |
|
LOQ |
0.66 µg |
27.25 ng |
|
%Recovery[%RSD][n=3] |
0.56– 0.96 |
0.34– 0.86 |
|
Ruggedness[%RSD] Analyst I[n=6] |
0.91 |
0.37 |
|
Analyst II[n=6] |
0.73 |
0.69 |
|
Precision[%RSD] Inter-Day[n =6] |
0.32– 0.48 |
1.13– 1.71 |
|
Intra-Day[n =6] |
0.20– 0.40 |
0.32– 1.08 |
|
Repeatability[n =6] |
0.68 |
0.39 |
3.9 Linearity
Table 3.1.2: Calibration Data for Linearity of Balofloxacin
|
Sr. No. |
Concentration of BLF (µg/mL) |
Peak Area (Mean±SD;n=6) |
%RSD |
|
|
2 |
306891 ±1061.33 |
0.34 |
|
|
4 |
482414 ±996.67 |
0.20 |
|
|
6 |
670048 ±2373.12 |
0.35 |
|
|
8 |
886579 ±1234.08 |
0.13 |
|
|
10 |
1067745 ±1810.76 |
0.16 |
|
|
12 |
1269769± 3491.43 |
0.27 |
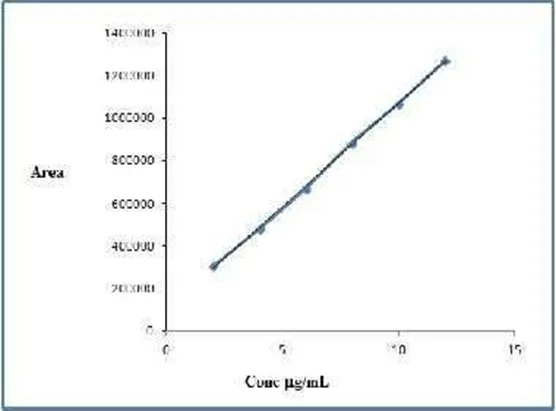
Figure3.1.2: Calibration Curve of Balofloxacin
CONCLUSION
A simple, rapid, accurate, precise, economical, and stability-indicating reverse-phase high-performance liquid chromatographic (RP-HPLC) method was successfully developed and validated for the quantitative estimation of balofloxacin in bulk drug and pharmaceutical tablet dosage forms. The chromatographic conditions were systematically optimized to achieve satisfactory peak resolution, acceptable retention time, excellent peak symmetry, and reproducible chromatographic performance. The developed analytical method was comprehensively validated according to the International Council for Harmonisation (ICH) guidelines for system suitability, specificity, linearity, accuracy, precision, robustness, ruggedness, limit of detection (LOD), and limit of quantification (LOQ), and all validation parameters complied with the recommended acceptance criteria. The proposed RP-HPLC method exhibited excellent linearity over the selected concentration range with a high correlation coefficient, while recovery studies confirmed the accuracy of the method and precision studies demonstrated excellent repeatability and intermediate precision with percentage relative standard deviation values well within acceptable limits. Robustness and ruggedness studies further established the reliability of the developed method under small deliberate variations in chromatographic conditions and routine laboratory environments. Forced degradation studies performed under acidic, alkaline, oxidative, thermal, photolytic, and neutral stress conditions demonstrated that the developed method effectively separated balofloxacin from its degradation products without interference, thereby confirming its stability-indicating capability. These findings indicate that the method is suitable for monitoring the stability of balofloxacin during pharmaceutical development, manufacturing, storage, and quality assurance studies. Overall, the validated RP-HPLC method is reliable, sensitive, cost-effective, and convenient for routine quantitative analysis of balofloxacin in bulk drug and pharmaceutical tablet dosage forms. Owing to its simplicity, short analysis time, excellent reproducibility, and compliance with ICH validation requirements, the proposed analytical method can be successfully employed for routine quality control testing, stability studies, regulatory submissions, and pharmaceutical research laboratories. Furthermore, the developed method may serve as a useful analytical tool for future formulation development and stability assessment of balofloxacin-containing pharmaceutical products.
REFERENCES


Kyatham Mamatha*, Gade Arunakumari, G. Hemalatha, Ankam Sindhuri, K. Jamuna, Development and Validation of a Stability-Indicating RP-HPLC Method for the Quantitative Estimation of Balofloxacin in Bulk Drug and Pharmaceutical Tablet Dosage Forms, Int. J. Med. Pharm. Sci., 2026, 2 (7), 734-746. https://doi.org/10.5281/zenodo.21379736
 10.5281/zenodo.21379736
10.5281/zenodo.21379736