
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
1Assistant Professor, Department of Pharmaceutical Chemistry, School of Pharmacy, Sai Nath University, Ranchi, Jharkhand, India. Email- joshishikhar236@gmail.com
2Assistant Professor, Department of Chemistry, S.S. Jain Subodh P.G. (Autonomous) College, Jaipur, Rajasthan, India
3Associate Professor, Department of Zoology, S.B.N.PG. College, Sikar Road, Jaipur.
4Master of Pharmacy (Pharmacology), Executive Assistant in Drug Regulatory Affairs at Venus Remedies Limited
HER2-positive breast cancer represents a biologically aggressive subtype characterised by overexpression or amplification of the human epidermal growth factor receptor-2 (HER2), which promotes tumour proliferation, metastasis, and poor clinical prognosis. The introduction of trastuzumab, a humanised monoclonal antibody targeting the extracellular domain of the HER2 receptor, has markedly improved survival outcomes and transformed the therapeutic landscape for patients with this disease. Nevertheless, intrinsic and acquired resistance to trastuzumab therapy remains a major clinical challenge, with a substantial proportion of patients demonstrating limited or transient responses to monotherapy. This systematic review evaluates emerging therapeutic strategies designed to enhance the clinical efficacy of trastuzumab through synergistic combination approaches. A structured literature search was conducted across major scientific databases to identify studies investigating trastuzumab-based combination therapies, including dual HER2 blockade with pertuzumab, antibody–drug conjugates such as ado-trastuzumab emtansine and trastuzumab deruxtecan, co-targeting strategies with epidermal growth factor receptor inhibitors such as nimotuzumab, and targeted nanotechnological platforms including trastuzumab-functionalized immunoliposomes and antibody–photosensitizer conjugates. The evidence indicates that these multimodal strategies improve therapeutic activity by inhibiting complementary signalling pathways, enhancing immune-mediated cytotoxicity, overcoming resistance mechanisms involving the PI3K/AKT/mTOR axis, and improving targeted drug delivery and tumour selectivity. Nanotechnology-based formulations further contribute by optimizing pharmacokinetics, reducing systemic toxicity, and enabling theranostic applications such as targeted imaging and photodynamic therapy. Collectively, these findings highlight the evolving paradigm of trastuzumab-based combination therapies as a central component of precision oncology in HER2-positive breast cancer. Continued translational and clinical investigations are required to refine patient selection, understand resistance mechanisms, and optimize therapeutic sequencing to achieve improved long-term outcomes for affected patients.
Cancer is currently the leading cause of mortality worldwide, with breast cancer representing one of the most significant contributors to global cancer burden It is the most commonly diagnosed malignancy among women and is associated with increasing incidence, elevated mortality rates, and substantial economic and social consequences [1], [2]. Breast cancer arises from the uncontrolled proliferation of cells in breast tissue and accounts for approximately 11% of all cancer diagnoses annually [3]. All women, irrespective of their racial or ethnic background, are at risk for breast cancer [4]. In 2012, approximately 1.7 million women were diagnosed with breast cancer, making it the most frequently diagnosed cancer in women in 140 of 184 countries. In high-income countries, breast cancer constitutes about 29% of all cancer cases and nearly 30% of cancer-related deaths. For instance, in the United States in 2009, approximately 192,000 new cases and 47,000 deaths were reported[5], [6]. Breast cancer is still the most common type of cancer in women worldwide, with a continually increasing global burden. An estimated 2.3 million new cases of female breast cancer were reported in 2020, accounting for 11.7% of all cancer cases worldwide, with 685,000 deaths, which accounted for 6.9% of all cancer-related mortality worldwide [7]. Projections for future cases show that by 2040, it is estimated that there may be 3 million or more cases of breast cancer, with 1 million or more deaths due to breast cancer, mainly caused by population growth and aging [8]. More recent global statistics for 2022 reported that there were 2.30 million new cases of breast cancer in women, with 0.67 million deaths, making it the leading cause of cancer mortality in women, whereas lung cancer, with 0.91 million cases, and cervical cancer, with 0.66 million cases, were the next most common cancers in women in terms of mortality [9]. Asia reported about 0.99 million new cases and 0.32 million deaths due to breast cancer at an age-standardized incidence rate of 34.3 per 100,000 and death rate of 10.5 per 100,000 in the year 2022, which is about 43% and 47% of the world’s cases and deaths, respectively [10]. The region with the highest mortality-to-incidence slopes was Africa, at 0.346 for individuals younger than 40 years old and 0.335 for individuals 40 years old or older, compared with Asia (0.085 and 0.208), Europe (0.002 and -0.014), and Northern America (-0.078 and -0.188). Therefore, this shows a higher fatality rate in regions with lower Human Development Index scores [10]. In different regions with different levels of Human Development Index scores, lower scores showed a higher mortality-to-incidence ratio, indicating global inequalities in breast cancer outcomes [11]. Lower Human Development Index scores showed higher mortality-to-incidence ratios in different regions, with Africa having a higher mortality-to-incidence slope in both age groups compared with Asia, Europe, and Northern America [12]. Prior research indicates that the mortality rate associated with breast cancer can be diminished through screening and early detection. Mammography, Breast Self-Examination, and Clinical Breast Examination are methods utilized for breast cancer screening [7]. Several barriers to breast cancer screening, including inadequate interactions with healthcare providers and challenges associated with the screening process [8]. Numerous risk factors for breast cancer development have been found, including age, menstruation, menopausal status, age at first live birth, family history, breastfeeding, genetic mutations, and lifestyle habits such as physical inactivity and a high-fat diet. The relationship between depression and the onset of breast cancer has been extensively subject to discussion. Additionally, Depression impacts the endocrine and immunological systems, potentially influencing the onset and course of cancer [9], [10].
Breast cancers are clinically classified into several categories [11]. The classifications includes ‘luminal A’ (estrogen receptor positive and/or progesterone receptor positive, human epidermal growth factor receptor 2 negative), ‘luminal B’ (estrogen receptor positive and/or progesterone receptor positive, human epidermal growth factor receptor 2 positive), ‘HER2 overexpressing’ (estrogen receptor negative, progesterone receptor negative, human epidermal growth factor receptor 2 positive), ‘basal-like’ (estrogen receptor negative, progesterone receptor negative, human epidermal growth factor receptor 2 negative, cytokeratin 5/6 positive and/or epidermal growth factor receptor positive), and ‘normal breast-like’ tumours [12].
HER receptors are linked to the progression of several human malignancies, notably breast cancer. The over activation of HER receptors promotes cancer progression. This excessive activation of HER receptors primarily results from overexpression caused by gene amplification; however, it may also arise from truncation of the extracellular domain, mutations in the kinase domain, or co-expression of HER receptor ligands [13], [14], [15]. EGFR overexpression occurs in 20–30% of breast cancer cases. A significant proportion of HER2-positive breast cancer cells also overexpress EGFR, while around 50% of triple-negative breast cancer cells exhibit EGFR overexpression. A high level of EGFR is often correlated with increased tumour growth and unfavourable clinical outcomes [16], [14], [17], [18]. ErbB2 overexpression is observed in 20–30% of breast and ovarian cancers [19], [14], [20], [21]. HER2 mutations are identified in approximately 1.6% of breast cancer patients [22]. Patients with breast cancers that overexpress ErbB2 exhibit a markedly reduced survival rate and a shorter time to relapse compared to those without ErbB2 overexpression [23], [24], [21]. Also, ErbB2 overexpression has been positively associated with lymph node metastasis in breast cancer [25]. ErbB3 overexpression is present in around 20% of breast tumours [26]. Overexpression primarily results from enhanced transcription [26], [27]. The sole overexpression of HER3 does not facilitate anchorage-independent growth; rather, its co-expression with HER2 significantly enhances cell proliferation [14], [28]. Unlike other HER receptors, HER4 has been associated with both oncogenic and tumour-suppressor functions (Fig. 1) [29].
Over 60 receptor tyrosine kinases (RTKs) have been found in the human genome [30]. Similar to other receptor tyrosine kinases (RTKs), HER receptors are single transmembrane proteins characterized by an N-terminal extracellular domain, a transmembrane helix, and a cytoplasmic domain [31]. The extracellular domain has four subdomains: the ligand-binding subdomains (domains I and III) and the receptor dimerization subdomains (domains II and IV). The intracellular domain consists of a tyrosine kinase domain and a C-terminal regulatory domain [32]. EGFR, a 170 kD monomeric polypeptide, serves as the typical member of the HER family receptors [33], [34]. EGFR and HER4 are fully operational receptor tyrosine kinases (RTKs) that may signal as both homo- and heterodimers upon ligand interaction, but the other two members, HER2 and HER3, exhibit distinct characteristics. HER2 is an orphan receptor devoid of a ligand, while HER3 exhibits an absence of kinase activity. Nevertheless, ligand-induced heterodimerization can fully activate all HER receptors to facilitate cell signalling [35], [36], [37], [38].

Fig. 1. The triggering of HER receptors and the subsequent downstream signalling cascades. Four HER receptor members engage with 11 ligands, leading to the formation and activation of 10 distinct homo- and heterodimers. Activated HER receptors stimulate several signalling cascades that influence various important biological outcomes.
In addition to EGF, eight additional ligands have been recognized to bind to and activate HER receptors. These ligands constitute the EGF family of peptide growth factors and are categorized into three groups according to their binding partners. EGF, epigen (EPG), amphiregulin (AR), and transforming growth factor (TGF) constitute a group that selectively interacts with EGFR. HB-EGF, epiregulin (EPR), and betacellulin (BTC) constitute the second group that interacts with both EGFR and HER4. The four neuregulin’s, namely NRG1, NRG2, NRG3, and NRG4, constitute the third group that interacts with HER4 [39], [40], [14], [41], [42]. NRG and NRG2 additionally attach to HER3. Each ligand individually influences the activation and signalling of the four HER receptors through its specific binding affinity and specificity [43]. Structural studies reveal that a total of ten homo- and heterodimers are formed by four HER receptors (Fig. 1) [44]. The conformations of these receptors are only available in two distinct forms: a tethered form and an extended form. The tethered form of the receptor is incapable of dimerization because the dimerization element is concealed. In the extended form, the dimerization elements of the receptor are completely accessible, facilitating the process of receptor dimerization [45], [46]. It is noteworthy and captivating to observe that HER2 extracellular domains exist in an extended conformation even in the absence of ligands. The interactions between subdomains I and III of the HER2 extracellular domains serve to directly stabilize HER2 in its extended conformation. The close interaction between subdomain I and III eliminates the possibility of a ligand occupying the space in between. Consequently, HER2 is inherently classified as a neglected receptor [47]. Consequently, HER2 sustains a ligand-independent and continuously activated conformation. HER2 can spontaneously form homodimers upon overexpression in cells, while other HER receptors show a preference for dimerization with HER2 [36]. The overexpression of HER2, unlike other HER receptors, results in cellular transformation and is associated with a poor prognosis in breast cancer [47]. In contrast, HER3 homodimer is generally considered non-functional because it lacks kinase activity. HER3 demonstrates markedly low kinase activity, roughly 1/1000th that of EGFR, suggesting the potential for functional HER3 homodimers [14]. All HER receptors activate through homo- or heterodimerization, phosphorylating C-terminal regulatory area tyrosine residues. Research, including extensive phosphor proteomic analyses, has identified more than 100 proteins that potentially interact with HER receptors [14], [48], [49], [50], [51], [52], [53]. The tyrosine phosphorylation residue mapping reveals some interesting aspects. EGFR and HER4 bind several downstream proteins. EGFR binds to Grb2, Shc, Src, PLC-1, Crk, Stat5, Ptp-2c, and SHP1. HER4 links Syk, RasA1, Abl, Crk, Vav2, and Grb2. HER2 and HER3 signalling pathways are limited and specialized. HER3 substantially activates the PI3K-Akt pathway due to its numerous phosphor tyrosine residues that bind to p85. On the other hand, HER2 primarily activates Erk pathways via Shc/Grb2. The heterodimerization of HER receptors activates more signalling cascades than the homodimer due to its unique binding to downstream signalling proteins. The HER2 homodimer primarily activates the Ras-ERK pathway based on binding selectivity, while the HER2-EGFR heterodimer may exhibit similar functionality. The HER2-HER3 heterodimer is potentially more effective than the HER2 homodimer or HER3 homodimer because it can activate all available receptors and both the PI3K-Akt and Ras-ERK pathways. Many studies have linked the PI3K-Akt pathway to HER2-HER3 signalling and HER2-positive breast cancer. HER2 is the most aggressive form of the disease, and approximately one in five women worldwide who are diagnosed with breast cancer will have HER2-positive breast cancer. In a patient-level meta-analysis of 2,518 patients with early-stage HER2-positive breast cancer in 11 studies with a median follow-up of 6.7 years, Villacampa et al. (2025) assessed the prognostic performance of the HER2DX genomic risk score in early-stage HER2-positive breast cancer patients. The results of the analysis indicated that patients in the low-risk category had a 6-year EFS of 93.6%, whereas those in the high-risk category had an EFS of 82.9%, showing an absolute difference of 10.7% and a multivariable stratified hazard ratio (HR) of 2.72. In addition, when assessed as a continuous variable, for each 10-unit increment in the HER2DX score, EFS decreased. These results were consistent across different clinical subgroups of disease and reflect the significant biological variability in HER2-positive disease, with activation of the PI3K-AKT pathway contributing to this variability [54]. Historically, surgery and chemotherapy were the mainstays of treatment for HER2-positive (HER2+) breast cancer, a subtype known for its aggressive behaviour, frequent diagnosis at advanced stages, and notable resistance to hormone therapies such as tamoxifen and to CMF (cyclophosphamide, methotrexate, 5-fluorouracil) chemotherapy. CMF demonstrated dose-dependent efficacy in HER2+ cases, indicating a potential advantage of high-dose chemotherapy; however, its overall effectiveness was still constrained [11], [12], [13], [14]. In the mid-1990s, taxanes (e.g., paclitaxel and docetaxel) demonstrated promising results both as monotherapy and in combination with anthracyclines, broadening the therapeutic options for HER2+ disease. Paik et al. (1998) demonstrated that the inclusion of anthracyclines in a treatment regimen of L-phenylalanine mustard and 5-FU markedly enhanced disease-free survival (DFS) in patients with HER2/ERBB2-positive breast cancer [15], [16].
Despite the development of multiple chemotherapy regimens with varying degrees of efficacy, uncertainty remained regarding the optimal regimen in terms of both safety and therapeutic benefit. Meanwhile, advancements in the understanding of HER2’s role as a prognostic and predictive biomarker continued to shape treatment strategies. A study by Tubbs et al. (2009) found that high HER2 gene overexpression was associated with poorer outcomes in patients receiving doxorubicin-based adjuvant therapy, underscoring the need for more targeted approaches. This evolving insight into HER2 gene amplification led to the development of HER2-targeted therapies, such as trastuzumab, which have since revolutionized treatment paradigms across various cancer types by offering more precise and effective intervention strategies [17], [18].
The 2019 Lasker-DeBakey Clinical Medical Research Award recognized Dennis J. Slamon, Axel Ullrich, and H. Michael Shepard for their groundbreaking invention of trastuzumab, the first-ever monoclonal antibody specifically targeting the IV extracellular subdomain of the oncogenic receptor HER2, and its advancement as a transformative treatment for women with HER2+ breast cancer. TZ was recognized by the Food and therapy Administration (FDA) in 1998 for the treatment of this particular breast cancer subgroup. Currently, eighty-five percent of HER2+ breast cancer patients are anticipated to survive for a minimum of ten years, representing a remarkable enhancement in patient outcomes [19].
Antibody-dependent cell-mediated cytotoxicity (ADCC) is identified as the extracellular mechanism associated with trastuzumab. The constant fraction (Fc) of the antibody (TZ), which is bound to its specific receptor on cancer cells, is recognized by natural killer cells via the CD16 receptor. This recognition activates innate immune cells to release lytic granules, including perforins and granzyme B, resulting in cancer cell death. This activity demonstrates that, in in vivo models, TZ activates the immune system, exerting a significant anti-tumour effect and enhancing the overall therapeutic efficacy of TZ. A comprehensive understanding of the role of ADCC in the immune response to TZ will facilitate the rational combination of these treatment modalities in the context of HER2+ BC [20], [21], [22], [23], [24], [25].
Numerous intracellular pathways have been suggested for the activity of trastuzumab; however, the evidence remains debatable. The suggested intracellular processes comprise: 1) The binding of TZ obstructs receptor dimerization and attenuates HER2-mediated signalling, thus decreasing cellular proliferation. The dimerization of HER2 involves phosphorylation, which generates docking sites for various downstream effectors, particularly those linked to the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB/AKT) pathway. The activation of this pathway occurs through the phosphorylation of AKT (p-AKT) and acts as a molecular marker for various human malignancies, as it regulates cell growth, survival, and motility [26]. Phosphorylation of AKT at Ser473 has been documented to facilitate breast cancer metastasis [27]. 2) Inhibition of the proteolytic cleavage of the extracellular domain of HER2, the binding to TZ not only obstructs HER2 dimerization and associated signalling but also promotes receptor degradation [28]. Moreover, TZ binding inhibits the cleavage of the HER2 extracellular domain (ECD) by metalloproteinases, leading to its release as sHER2, the soluble variant, and the formation of a membrane-bound phosphorylated p95 that maintains the ability to activate signal transduction pathways. (Figure 2) [20]. 3) Inhibition of DNA repair mechanisms. Chemotherapy and radiotherapy induce DNA damage in cancer cells, which may be mitigated by DNA repair mechanisms. Preliminary research indicates that trastuzumab partially inhibits the repair of damaged DNA [29], [30]. 4) Inhibition of angiogenesis. Malignant cells stimulate angiogenesis to facilitate tumour proliferation. Trastuzumab demonstrated the ability to decrease angiogenesis in a preclinical mouse xenograft tumour model [31], [32].

Figure 2. Illustrates the structure and function of the HER2 receptor and its suppression by trastuzumab. (A–C) Depict the receptor's construction, activation, internalization, recycling, and shedding; (D) Demonstrates the interaction of the monoclonal antibody trastuzumab (TZ) with HER2 at a location inside the IV extracellular domain of the receptor, hindering its functionality.
Since its approval in 1998, trastuzumab has transformed the treatment landscape for metastatic HER2-positive breast cancer, significantly improving clinical outcomes and overall survival rates. Despite its groundbreaking success, the clinical utility of trastuzumab is hampered by several critical limitations. The efficacy of trastuzumab, a monoclonal antibody targeting the HER2 receptor, can be compromised by the expression of other members of the HER family. This occurs due to the presence of redundant ligands and receptors, which facilitate alternative dimerization patterns and activate downstream signalling pathways, thereby contributing to therapeutic resistance [33], [34]. Approximately 50% of patients respond to trastuzumab therapy, yet nearly 40% exhibit primary resistance, failing to respond from the outset [35], [36], [37], [38]. Additional challenges associated with trastuzumab monotherapy include adverse side effects, safety concerns, limited circulation time, suboptimal efficacy, patient compliance issues, and challenges in drug administration. These limitations have prompted extensive research efforts focused on developing more effective and durable therapeutic strategies to overcome resistance and enhance treatment outcomes in HER2-positive breast cancer.
Several therapeutic strategies have been developed to overcome trastuzumab resistance and enhance therapeutic efficacy. These include dual HER2 blockade with pertuzumab, an antibody targeting a different epitope of HER2; small-molecule tyrosine kinase inhibitors such as lapatinib; antibody-drug conjugates (ADCs) like ado-trastuzumab emtansine (T-DM1) and trastuzumab deruxtecan (T-DXd); and combination regimens involving chemotherapy, targeted inhibitors of the PI3K/AKT/mTOR pathway, or even anti-EGFR agents like nimotuzumab. These combinatorial strategies aim to provide a more complete blockade of HER2-driven oncogenic signalling and address the multifactorial nature of resistance. Given the expanding landscape of trastuzumab-based combination therapies, it is crucial to systematically assess the efficacy, safety, pharmacological mechanisms, and clinical outcomes associated with these regimens. A comprehensive synthesis of available evidence is essential to inform clinical decision-making and identify knowledge gaps for future research. This systematic review attempts to assess the existing evidence regarding trastuzumab-based combination strategies for treating HER2-positive breast cancer. It aims to: Examine the clinical effectiveness and survival results linked to combination therapies that include trastuzumab. Examine the mechanisms that contribute to trastuzumab resistance and the ways in which combination therapies mitigate these issues. Analyse the pharmacodynamic and pharmacokinetic characteristics of trastuzumab across various therapeutic settings. Evaluate the safety profiles and tolerability of trastuzumab-based combinations in both early and advanced stages of disease. This review synthesizes current data to elucidate the evolving role of trastuzumab-based therapies in HER2-positive breast cancer, while also highlighting promising strategies to overcome therapeutic resistance. A structured literature search was conducted to identify studies on the evaluation of advancements in the therapy involving trastuzumab and synergistic therapy strategies in HER2-positive breast cancer. The relevant literature was collected from various scientific literature databases. The literature search was carried out using various keywords such as ‘trastuzumab,’ ‘HER2-positive breast cancer,’ ‘adjuvant therapy,’ ‘combination therapy,’ ‘synergistic therapy,’ ‘HER2 targeted therapy,’ ‘antibody-dependent cellular cytotoxicity,’ ‘trastuzumab resistance,’ and ‘HER2 signaling inhibition.’ Boolean logical operators AND and OR were used in the literature search strategy to narrow down the search results and focus on the literature involving combination therapy and emerging trends in the therapy involving trastuzumab in the management of HER2-positive breast cancer. The relevant literature involving adjuvant therapy and synergistic therapy strategies was considered for the study.
Survival and Prognostic Factors in HER2-Positive Breast Cancer Post-Trastuzumab Therapy
Trastuzumab has significantly improved outcomes for patients with HER2-positive breast cancer, resistance to therapy remains a major clinical concern. Various molecular and clinical factors have been proposed as potential predictors of therapeutic response or resistance. These factors are increasingly being recognized as crucial prognostic indicators and are the focus of numerous clinical trials in both early-stage and advanced disease settings [39]. Trastuzumab is a humanized IgG1 monoclonal antibody that binds to the extracellular domain IV of the HER2 receptor protein, also known as ERBB2. The structure is similar to the usual IgG1 structure with an approximate molecular weight of 150 kDa and is composed of two heavy chains and two light chains with conserved disulfide bonds and an Fc portion that can mediate effector functions such as antibody-dependent cellular cytotoxicity. The monoclonal antibody has high antigen binding capacity with low nanomolar affinity, with the KD being 8.2 nM for unconjugated trastuzumab in BT-474 cells; however, this affinity may slightly change after the conjugation process [40]. Analytical tests have revealed that trastuzumab has physicochemical stability when left at room temperature (20-25°C) for 12 hours, with no changes observed in the UV-Vis spectrum, secondary structure using FTIR, banding pattern using SDS-PAGE, and aggregation using size exclusion chromatography; however, the HER2 binding affinity of ^111In-BzDTPA-trastuzumab is stable with KD values between 2.2 and 4.4 nM and in vivo tumor uptake [41]. For radiotheranostic applications, conjugates such as NOTA-trastuzumab, with approximately 2.9 chelators per antibody, showed immunoreactivity close to 69% and had low nanomolar binding affinity around 2.1 nM, with high tumor uptake in HER2-positive xenograft models, suggesting that structural modification can change hydrodynamic characteristics while maintaining target-binding and functional activities [40]. The therapeutic activity of trastuzumab in HER2-overexpressing breast cancer is primarily due to its ability to interfere with oncogenic HER2 signaling and induce immunologically mediated destruction of tumor cells [42]. Trastuzumab targets the extracellular domain of the HER2 receptor, which in turn reduces phosphorylation of the HER2 receptor and subsequently reduces activity of the PI3K/AKT and MAPK/ERK pathways, which are involved in cellular proliferation and apoptosis, respectively; combinations of therapies that further reduce HER2 signaling may augment this effect, as seen with PEG-IFN-α1b, which enhanced inhibition of HER2, AKT, and ERK signaling and had synergistic activity with trastuzumab in xenograft models [42]. However, these resistance mechanisms may preserve HER2 signaling activity by activating other signaling regulators to promote tumor growth, underscoring the importance of HER2 signaling inhibition in trastuzumab treatment of breast cancer [43]. Trastuzumab is also known to exert antitumor effects through immune mechanisms in which its Fc region is involved in interactions with immune effector cells, especially natural killer (NK) cells, to exert antibody-dependent cellular cytotoxicity (ADCC) [42]. The activation of NK cells has been shown to enhance the killing of tumor cells, and experimental studies have shown that enhanced NK cell activity is associated with the effectiveness of trastuzumab, and depletion of NK cells has been shown to eliminate the observed synergistic antitumor effects of trastuzumab, thus validating ADCC as a significant mechanism of action of the drug [42]. The current evidence suggests that immune mechanisms are mediated through interactions with Fc gamma receptors, especially CD16A on NK cells, which are critical in the antitumor effects of trastuzumab and are of significant importance as targets for approaches aimed at improving the effectiveness of the drug and overcoming resistance to therapy [43]. Drug resistance is characterized by either a lack of positive therapeutic response (intrinsic resistance) or disease progression following an initial clinical benefit (acquired resistance). Intrinsic resistance mechanisms to trastuzumab emerge prior to the initiation of therapy. Many are associated with an inactive target receptor, such as truncated HER2 receptors that lack the extracellular trastuzumab-binding domain, or modifications of downstream components in the PI3K/Akt/mTOR signalling pathway. Acquired resistance primarily results from modifications at the target signalling level and requires an active target receptor. The upregulation of additional TKRs or their ligands is included in this category [40]. Although certain mechanisms have been identified in both groups. The various mechanisms are categorized as follows (Fig. 3). Resistance may develop because to the modified HER2 expression status of the cancer cells [41], [42]. Resistance may also emerge due to modifications in HER2 molecule structures, such as proteolytic cutting of the HER2 extracellular domain, which inhibits the binding of trastuzumab to the shortened yet constitutively active HER2 [43], [44], [45], [46]. Activation of other HER receptors, such as EGFR, compensate for the diminished HER2 signalling resulting from trastuzumab inhibition, or HER2 may be activated by a mechanism that is resistant to trastuzumab [47], [48], [49]. Mutations causing constitutive activation of downstream signalling pathways are a significant mechanism of trastuzumab resistance. The most notable case is the constitutive activation of the PI3K-Akt-mTor pathway resulting from a gain-of-function mutation in PI3K and the loss-of-function mutation in PTEN [50], [51], [52], [53], [54], [55]. Additional pathways have been documented, including FC receptor polymorphism, miRNAs and mucin 4 expression caused by TNF [56], [57], [58]. In a preclinical study by Lin et al. (2026), a trastuzumab-resistant SK-BR-3 breast cancer cell line (SK-BR-3R) was derived from SK-BR-3 human breast cancer cells and found to have a resistance index of 13.79 compared with its parental SK-BR-3 cells. In this study, SK-BR-3R showed a significant upregulation in HER2 expression as well as in phosphorylated PI3K (p-PI3K). In vivo SPECT/CT imaging showed a higher uptake of the HER2-targeting probe 99mTc-HP-Ark2 in SK-BR-3R tumor models compared with tumor models from parental SK-BR-3 cells. Inhibition of PI3K activity with LY294002 resulted in decreased expression of HER2 as well as decreased uptake of 99mTc-HP-Ark2 in SK-BR-3R cells, along with restored sensitivity to trastuzumab. Furthermore, dual therapy with LY294002 and trastuzumab showed a more potent inhibitory effect on HER2 expression than either therapy alone [59].
The HER2/HER3 heterodimer-mediated activation of the PI3K-Akt-mTor signalling pathway is regarded as the principal mechanism driving breast cancer development, with its constitutive activation recognized as a significant contributor to trastuzumab resistance. Consequently, the combined inhibition of both HER2 and the PI3K-Akt-mTor pathway has been investigated in order to overcome trastuzumab resistance [59], [60]. Numerous studies have shown that further suppression of the PI3K-Akt-mTor pathway may surmount trastuzumab resistance in HER2-positive breast tumours [61], [62], [63], [64]. Furthermore, prognostic biomarkers such as PTEN loss, PIK3CA mutations, and p95HER2 expression are being increasingly validated in clinical settings. These biomarkers may facilitate risk stratification and guide personalized treatment strategies in HER2-positive breast cancer. In a meta-analysis conducted by Peng et al. (2022), data from five randomized trials of 1,548 patients were assessed to investigate the effectiveness of the addition of PI3K/Akt/mTOR pathway inhibitors to trastuzumab in the treatment of HER2-positive breast cancer. The results of the analysis indicated that the addition of a pathway inhibitor significantly improves PFS with a hazard ratio of 0.82 (95% CI, 0.76–0.90), and this is more pronounced in hormone receptor-negative breast cancer with a hazard ratio of 0.73 (95% CI, 0.58–0.93). These results are in line with the pathway in which activation of the PI3K-Akt-mTOR signaling pathway is known to cause resistance to trastuzumab therapy [65].

Fig. 3: Mechanism of trastizumab resistance
Abbreviations: Akt, protein kinase B; Cdk 2/4, cyclin-dependent kinase 2/4; E2, estradiol; ER, estrogen receptor; IGF1R, insulin-like growth factor I receptor; HER, human epidermal growth factor receptor; MAPK, Mitogen-activated protein kinases; mTOR, mammalian target of rapamycin; P, phosphorylation; PI3K, phosphatidylinositol 3’-kinase; PI3Kmut, mutated phosphatidylinositol 3’-kinase; PTEN, phosphatase and tensin homolog.
Advances in Trastuzumab-Based Therapy: Adjuvant and Synergistic Strategies in HER2-Positive Breast Cancer
Synergistic HER2 Inhibition with Trastuzumab and Pertuzumab
Pertuzumab, a fully humanized recombinant monoclonal antibody, constitutes a novel class of agents that inhibit HER2 dimerization by binding to a distinct epitope of the HER2 receptor. It has shown synergistic effects with trastuzumab, thereby enhancing anti-tumour activity and improving survival outcomes. In the adjuvant setting, dual HER2 blockade using trastuzumab and pertuzumab, especially when paired with chemotherapy, has proven to be a promising approach for patients with high-risk, early-stage HER2-positive breast cancer [65], [66]. Multiple mechanisms have been suggested to elucidate the reported synergism of trastuzumab and pertuzumab in the treatment of HER2-positive cancers, including breast, ovarian, non-small cell lung, and gastric cancers [67], [68], [69], [70]. These processes consist of (1) The synergistic effect arising from the distinct roles of these two antibodies in targeting HER2-positive cancer cells [71]. (2) Synergism results from the composition-independent inhibitory effects of the combination of the two antibodies across a wide range of HER2/HER3 compositions [72], and (3) The synergistic effect of trastuzumab and pertuzumab is partially due to their enhanced binding affinity for the HER2 molecule, which arises from the cooperative interactions between the two antibodies [73]. The initial process can be verified by experimental data, while the subsequent two are exclusively based on computational models. Therefore, the mechanism will be discussed in more detail. Trastuzumab and pertuzumab have been assigned various roles, which may illustrate the synergistic effects of the two antibodies. A widely accepted hypothesis posits that trastuzumab inhibits the homodimerization of HER2 and the associated signalling pathways activated by HER2 homodimers, whereas pertuzumab primarily prevents the heterodimerization of HER2 with EGFR, HER3, and HER4, along with the downstream signalling pathways triggered by HER2 heterodimers (Figure 4) [74], [71]. This notion has been verified by certain research results [75]. Trastuzumab, rather than pertuzumab, has been reported to interfere with ligand-independent signalling facilitated by the HER2 homodimer. Trastuzumab was demonstrated to disrupt ligand-independent interactions between HER2 and HER3 [76]. Pertuzumab inhibits ligand-mediated dimerization of HER2 with HER3 [75], [77], [78]. In a study by Kang et al. (2025), the interaction between pertuzumab and trastuzumab deruxtecan (T-DXd) was examined in HER2-overexpressing models with varying HER2-HER3 dimerization activity. The dual treatment demonstrated greater growth inhibition in NCI-N87 cells, which have high HER2-HER3 activity, but no synergy was observed in OE19 cells with low HER2-HER3 activity. Neuregulin-1 decreased the activity of T-DXd, but pertuzumab partially restored the effect of T-DXd in NCI-N87 cells. The dual treatment also demonstrated greater inhibition of tumor growth in vivo in NCI-N87 cells compared to monotherapy, confirming inhibition of ligand-dependent HER2-HER3 signaling [79].

Fig. 4: Proposed models demonstrating the synergistic effects of trastuzumab and pertuzumab. (A) preferred theory proposes the unique effects of trastuzumab and pertuzumab on HER2 homo- and heterodimers, irrespective of ligand presence. (B) Hypothesis derived from computational analysis indicating synergistic interactions between the two antibodies.
The idea that the synergy is due to these two antibodies' different functions in targeting HER2-positive cancer cells may still dominate as more data supports it. Pertuzumab may impair HER2-mediated cell-signalling cascades, particularly those initiated by heregulin-induced HER2/HER3 heterodimerization. Pertuzumab inhibits HER2 heterodimer formation and intracellular signalling downstream of it, according to all available studies. Further evidence suggests that pertuzumab disrupts heregulin-induced HER2/HER3 heterodimers and activates the PI3K/Akt pathway downstream [75], [77], [79]. Trastuzumab may function downstream of HER2 via non-canonical mechanisms. Trastuzumab routinely exhibits high ADCC [80], [81], [25], [24], [22], [82], [83]. Trastuzumab inhibits HER2 endocytosis/downregulation [84], [20], DNA repair [29], extracellular domain proteolytic cleavage [85], and angiogenesis [86], [87]. Trastuzumab, but not pertuzumab, has been found to suppress apoptosis and enhance reactive oxygen species generation in human cardiomyocytes by disrupting HER2 signalling [88]. Computational and molecular modeling studies have provided mechanistic insights into the synergistic action of trastuzumab and pertuzumab in targeting HER2-positive cancers. These studies reveal that trastuzumab and pertuzumab differentially inhibit HER receptor-mediated signalling based on receptor dimerization patterns, with pertuzumab showing stronger binding affinity to HER2/HER3 heterodimers due to lower ΔG binding and higher interfacial contact (IC) values. Additionally, modeling suggests that the synergy may arise from cooperative binding, where the antibodies colocalize on the HER2 extracellular domain, enhancing overall binding affinity and inhibiting receptor dimerization. While some experimental findings support enhanced dual-antibody binding, other studies contest the cooperative effect, indicating no significant enhancement in binding kinetics when both antibodies are used together. In a study carried out by Cruz et al. (2023), the authors used a combination of in vitro and in silico approaches to investigate the binding properties of pertuzumab and trastuzumab with HER2. In silico studies using the PRODIGY method showed that pertuzumab had slightly stronger HER2-binding properties, indicated by a better predicted binding free energy, mainly because of the higher interfacial contact scores. This was also observed in vitro. Structural analysis showed that pertuzumab IgG had a greater ability to form bivalent complexes with HER2, whereas trastuzumab had a greater ability to form monovalent complexes because of steric constraints, suggesting differences in their effects on dimerization and regulation of receptor signaling [89]. The APHINITY trial was a pivotal phase III randomized, double-blind, placebo-controlled study evaluating the efficacy of adjuvant pertuzumab in combination with trastuzumab and chemotherapy in patients with early-stage HER2-positive operable breast cancer. A total of 4,805 women with either node-positive disease or high-risk node-negative disease were enrolled. Participants were randomized to receive either pertuzumab or placebo in combination with trastuzumab for one year, following standard chemotherapy regimens. The chemotherapy backbone included either anthracycline/taxane-based regimens or taxane plus carboplatin protocols. The primary endpoint of the trial was invasive disease-free survival (iDFS). At three years, the iDFS was 94.1% in the pertuzumab group compared to 93.2% in the placebo group, reflecting a modest absolute benefit of 0.9%. However, in the node-positive subgroup, the iDFS improvement was more pronounced—92.0% vs. 90.2%, with a statistically significant p-value of 0.02. Among hormone receptor-positive patients, the iDFS benefit was 2.0% (94.8% vs. 92.8%), although this difference did not reach statistical significance. These findings suggest that while the overall benefit of adding pertuzumab is modest, it may be clinically meaningful for patients with high-risk features, particularly lymph node involvement, where the risk of recurrence is higher [89]. Both trastuzumab and pertuzumab are IgG1 monoclonal antibodies administered via intravenous infusion and exhibit nonlinear pharmacokinetics influenced by target-mediated drug disposition. Pertuzumab has a terminal half-life of approximately 18 days, with steady-state concentrations typically achieved by the third cycle of every-three-week dosing. The recommended regimen consists of an 840 mg loading dose, followed by 420 mg every 3 weeks. Its volume of distribution approximates 3.0–5.0 L, and clearance (CL) is reported at 0.235 L/day, with minimal renal excretion [90], [91]. Trastuzumab, administered as a loading dose of 8 mg/kg followed by 6 mg/kg every 3 weeks, has a terminal half-life ranging from 18 to 25 days, and a steady-state is reached by week 20. Trastuzumab’s clearance ranges between 0.225–0.399 L/day, with volume of distribution around 3–7 L [90], [91], [92], [93]. The addition of pertuzumab to standard trastuzumab-based therapy in the adjuvant setting was associated with an increased incidence of adverse events, the most notable being grade ≥3 diarrhea, which occurred in 9.8% of patients in the pertuzumab group compared to 3.7% in the placebo arm. Other adverse effects were consistent with the established safety profiles of HER2-targeted monoclonal antibodies and were generally manageable with supportive care [94], [95]. Further support for dual HER2 blockade arises from key neoadjuvant trials such as TRYPHAENA and BERENICE, both of which demonstrated high pathological complete response (pCR) rates and acceptable cardiac safety when pertuzumab was added to trastuzumab and chemotherapy [89], [96]. Collectively, these data justify the continued use of trastuzumab and pertuzumab in combination for one year as part of a comprehensive HER2-targeted treatment strategy in selected high-risk patients, with ongoing research needed to refine patient selection, optimize sequencing with novel agents, and reduce treatment-associated toxicities.
Ado-Trastuzumab Emtansine Adjuvant HER2 Breast Cancer Treatment
Ado-trastuzumab emtansine (T-DM1) represents a major advancement in HER2-targeted therapy, functioning as the first antibody–drug conjugate (ADC) specifically developed for HER2-positive breast cancer. It consists of the humanized anti-HER2 IgG1 monoclonal antibody trastuzumab covalently linked to the cytotoxic agent DM1, a maytansinoid that inhibits microtubule assembly, via a non-reducible thioether linker known as MCC (4-[N-maleimidomethyl] cyclohexane-1-carboxylate). Each T-DM1 molecule contains an average of 3.5 DM1 molecules linked to a trastuzumab antibody [97]. On February 22, 2013, the Food and Drug Administration (FDA) first approved T-DM1 for the treatment of HER2-positive metastatic breast cancer patients who had already undergone trastuzumab plus a taxane, either alone or in combination [98], [99]. This drug combination works by triggering receptor-mediated endocytosis, which leads to the internalization of the HER2-T-DM1 complex, which is achieved via T-DM1 binding to HER2 [100], [101]. Proteolytic breakdown of the antibody component of T-DM1 within the lysosome is the sole mechanism by which active DM1 release occurs, as the non-reducible linker remains stable in both circulation and the tumour microenvironment [102]. When DM1-containing metabolites exit the lysosome, they obstruct microtubule assembly, which in turn causes cell death (Fig. 5) [103]. In a study presented by Delgado et al. (2021), the efficacy of adjuvant treatment with ado-trastuzumab emtansine (T-DM1) was assessed in patients with early breast cancer who were positive for HER2 and had residual disease after neoadjuvant therapy. In a study of 1,486 participants in the KATHERINE trial, the 3-year invasive disease-free survival rate was 88.3% in the T-DM1 group, compared with 77.0% in the group of patients who received trastuzumab alone, with a hazard ratio of 0.50. However, more adverse effects, including hepatotoxicity, thrombocytopenia, peripheral neuropathy, hemorrhage, and pulmonary toxicity, were reported in the group of patients who received T-DM1 [104].
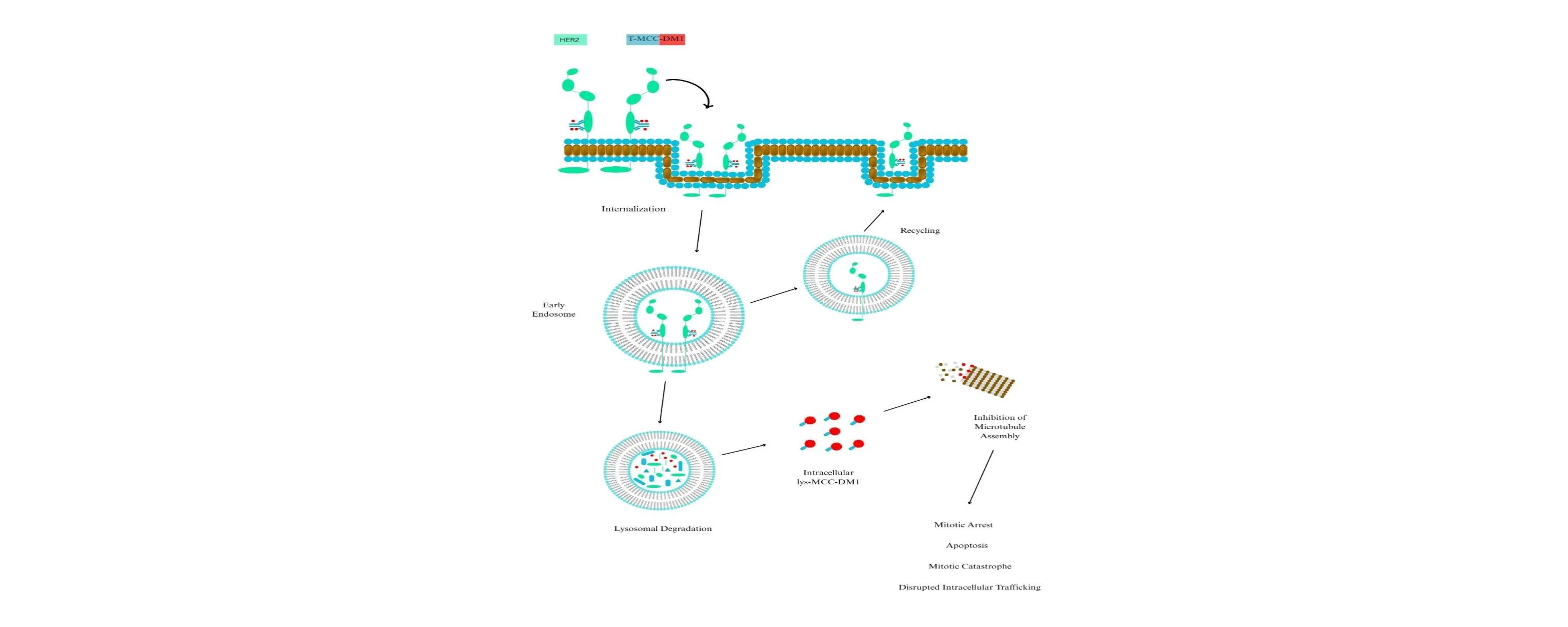
Fig. 5: Intracellular trafficking of trastuzumab emtansine (T-DM1) is a critical process in its therapeutic efficacy. The binding of T-DM1 to the human epidermal growth factor receptor-2 (HER2) on the plasma membrane is succeeded by the internalization of the HER2-T-DM1 complex through receptor-mediated endocytosis. Internalized endocytic vesicles give rise to early endosomes. Early endosomes can either recycle their load back to the cell membrane or mature into lysosomes. The release of DM1 results from the proteolytic degradation of the antibody component of T-DM1 within lysosomes. Intracellular lysine (lys)-MCC-DM1 inhibits microtubule assembly, resulting in mitotic arrest, apoptosis, mitotic catastrophe, and disrupted intracellular trafficking. MCC, a non-reducible thioether linker.
Trastuzumab and T-DM1 inhibit HER2 receptor signalling, promote antibody-dependent cell-mediated cytotoxicity, and inhibit the shedding of the extracellular domain of HER2 [104], [105]. The anti-tumour efficacy of DM1 surpasses that of trastuzumab; however, the effects of trastuzumab remain important, particularly in scenarios where target cells do not undergo rapid apoptotic death induced by DM1 [104]. Active DM1 metabolites disrupt the microtubule networks in target cells, leading to cell cycle arrest at the G2-M phase and subsequent apoptotic cell death [105], [106]. Secondly, prolonged treatment of breast cancer xenografts with T-DM1 induced both apoptosis and mitotic catastrophe, the latter defined by the presence of cells displaying abnormal mitotic shapes and large multinucleated structures [105]. Third, interruption of intracellular flow mediated by the microtubule network may occur. Microtubule-targeting drugs often disrupt intracellular transport via microtubules. Prolonged administration of T-DM1, in contrast to trastuzumab, led to impaired intracellular trafficking of HER2 in a preclinical breast cancer model [105], [107], [108]. Impaired intracellular transport may represent a crucial mechanism of action for T-DM1, particularly in non-proliferating cells. From a pharmacokinetic (PK) standpoint, T-DM1 demonstrates linear, time-independent kinetics when administered intravenously at the recommended dosage of 3.6 mg/kg every three weeks. The mean maximum serum concentration (C_max) of T-DM1 reaches approximately 80 µg/mL, and the area under the concentration-time curve extrapolated to infinity (AUC_inf) ranges between 400 and 500 µg·day/mL [107]. The terminal elimination half-life is approximately 3.5 to 4 days, which is consistent with the PK properties observed for other IgG1 monoclonal antibodies. T-DM1 has a relatively low steady-state volume of distribution (Vd), around 3–4 liters, indicating that the drug predominantly remains within the vascular and interstitial compartments. Systemic clearance (CL) is estimated at 0.68 L/day, with no significant accumulation over repeated dosing cycles [109].
T-DM1 has demonstrated substantial antitumour efficacy in both preclinical studies and clinical trials. Compared to trastuzumab, T-DM1 exhibits superior activity in trastuzumab-sensitive breast cancer cell lines and tumour xenograft models. Notably, it also retains efficacy in in vitro and in vivo models of trastuzumab-resistant breast cancer [105], [106]. A pivotal Phase III clinical trial, EMILIA, evaluated the efficacy and safety of T-DM1 in 991 patients with previously treated locally advanced or metastatic HER2-positive breast cancer. Patients received T-DM1 at a dose of 3.6 mg/kg intravenously every three weeks. The median progression-free survival (PFS) was 9.6 months, significantly longer than that observed with trastuzumab-based therapy. Adverse events observed with T-DM1 included thrombocytopenia, elevated liver enzymes, and fatigue. Although thrombocytopenia was more frequent in the T-DM1 arm, serious adverse events were fewer overall compared to standard chemotherapy regimens [110]. These clinical outcomes contributed to the regulatory approval of T-DM1 and its integration into treatment guidelines for HER2-positive MBC, especially for patients previously treated with trastuzumab and chemotherapy. Overall, T-DM1 exemplifies the evolution of targeted therapy in oncology by combining the specificity of monoclonal antibodies with the cytotoxic potency of chemotherapy, offering a well-tolerated and effective therapeutic option for patients with HER2-positive breast cancer.
Trastuzumab Deruxtecan adjuvant therapy
Trastuzumab deruxtecan (T-DXd) is a potential new antibody-drug conjugate for the treatment of patients with HER2-positive and HER2-low breast cancer. Despite substantial advancements in treatment outcomes for HER2-positive breast cancer patients in recent years, numerous patients continue to experience disease progression on existing regimens. Consequently, innovative agents like T-DXd are of significant interest. This study evaluates the efficacy and safety profile of T-DXd in treating breast cancer patients [111].
T-DXd is an antibody-drug conjugate (ADC) consisting of a humanized anti-HER2 IgG1 monoclonal antibody, identical in amino acid sequence to trastuzumab, covalently linked to a topoisomerase I inhibitor payload (DXd, a derivative of the camptothecin analogue exatecan) via a cleavable tetrapeptide-based linker. T-DXd demonstrates an increased Drug-to-Antibody Ratio (DAR) [112], [113], [114]. The elevated DAR facilitates the delivery of a concentrated cytotoxic payload, DXd, to the target cells [115]. The payload of T-DXd (DXd) induces antibody-dependent cellular cytotoxicity (ADCC) through two mechanisms: the inhibition of topoisomerase I, affecting topoisomerase I–DNA complexes, and antibody binding to FcgRIII regions on immune effector cells [115]. The inhibition of the topoisomerase I-DNA complex results in the suppression of DNA replication, cell cycle arrest, and apoptosis in tumour cells [116], [114], [114]. Antibody binding leads to a downregulation of phosphorylated Akt, which is part of a signal transduction pathway that promotes cell survival and growth, and an upregulation of the cyclin-dependent kinase inhibitor p27, ultimately resulting in the inhibition of cellular proliferation [115]. In vitro pharmacokinetic studies demonstrated that the DXd payload is a potent DNA topoisomerase I inhibitor, with an IC₅₀ of 0.31 μmol/L. T-DXd exhibited strong antitumour activity in HER2-positive cell lines and in vivo models of HER2-positive and HER2-low breast cancer [115], [114]. Notably, T-DXd retained efficacy in T-DM1-resistant tumour cells across both high and low HER2 expression levels [115]. The membrane-permeable nature of DXd facilitates a bystander killing effect, allowing the cytotoxic payload to exert antitumour activity in adjacent tumour cells independent of HER2 expression [117]. In immunocompetent mouse models bearing human HER2-expressing tumours, T-DXd also enhanced antitumour immunity, evidenced by upregulation of dendritic cell markers, increased MHC class I expression in tumour cells, and rejection of rechallenged tumours via adaptive immune responses [118]. A population pharmacokinetic analysis demonstrated that following intravenous administration of trastuzumab deruxtecan (T-DXd) at the recommended dose of 5.4 mg/kg once every 3 weeks for HER2-positive metastatic breast cancer, the geometric mean maximum plasma concentrations were 122 μg/mL for T-DXd and 4.4 ng/mL for the released cytotoxic payload, DXd [119], [120]. In an open-label, phase 1 trial comprising dose-escalation and dose-expansion cohorts, trastuzumab deruxtecan (T-DXd) demonstrated antitumour activity and a manageable safety profile in heavily pretreated patients with HER2-expressing metastatic breast cancer, including those with HER2-positive T-DM1-resistant and HER2-low disease [121]. The dose-escalation phase established 5.4 mg/kg and 6.4 mg/kg as the recommended doses [120]. The dose-expansion cohort of HER2-positive metastatic breast cancer demonstrated a confirmed objective response rate (ORR) of 59.5% (95% CI, 49.7–68.7), with a median duration of response (DOR) of 20.7 months and a median progression-free survival (PFS) of 22.1 months [119]. Among patients with heavily pretreated HER2-low metastatic breast cancer in the phase 1b dose-expansion cohort, independent central review reported a confirmed ORR of 37.0% (95% CI, 24.3–51.3) and a median DOR of 10.4 months [121].
DESTINY-Breast01 was a two-part, open-label, single-group, multicentre phase 2 trial assessing trastuzumab deruxtecan (T-DXd) in patients with HER2-positive metastatic breast cancer who had previously received two or more anti-HER2 therapies, including T-DM1 [122]. Study focused on pharmacokinetics and dose finding; based on safety and efficacy, a recommended dose of 5.4 mg/kg every 3 weeks was established [122]. In the most recent analysis (median follow-up: 26.5 months), T-DXd demonstrated an objective response rate (ORR) of 62.0% (95% CI: 54.5–69.0), median duration of response (DOR) of 18.2 months (95% CI: 15.0–not evaluable), median progression-free survival (PFS) of 19.4 months (95% CI: 14.1–25.0), and median overall survival (OS) of 29.1 months (95% CI: 24.6–36.1) [123]. The safety profile remained consistent with earlier findings. Grade ≥3 treatment-emergent adverse events (TEAEs) occurred in 53.8% of patients, with the most common being nausea, fatigue, alopecia, vomiting, constipation, decreased appetite, diarrhoea, and anaemia. Interstitial lung disease (ILD) was reported in 15.8% of patients, with 2.7% resulting in death, as confirmed by independent adjudication [122], [123]. In a study by José Baselga Cortés et al. (2024), which was reported on, updated results from the DESTINY-Breast03 trial investigated trastuzumab deruxtecan (T-DXd) in HER2-positive metastatic breast cancer previously treated with trastuzumab in combination with a taxane. T-DXd showed a significant improvement in progression-free survival over T-DM1, with a median progression-free survival of 29.0 months versus 7.2 months, along with a 36-month progression-free survival rate of 45.7% vs 12.4%. In addition, overall survival was also in favor of T-DXd (52.6 months vs 42.7 months) [124]. T-DXd represents a significant advancement in the treatment of HER2-positive and HER2-low breast cancer, offering efficacy in patients previously treated with T-DM1 and demonstrating activity in HER2-low tumours. Its dual mechanism, potent cytotoxic payload, and bystander effect support its use in resistant disease settings. These data have led to the global regulatory approval of T-DXd for HER2-positive unresectable and/or metastatic breast cancer, with ongoing trials expanding its potential role in earlier disease settings.
Synergistic mTOR Inhibition via Trastuzumab-Functionalized Immunoliposomes: A Nanotherapeutic Strategy for HER2-Positive Breast Cancer
The integration of nanotechnology with targeted therapies, such as the encapsulation of rapamycin in trastuzumab-functionalized immunoliposomes, presents a novel strategy aimed at enhancing efficacy while reducing side effects. This study evaluates the therapeutic potential, physicochemical characteristics, and preclinical performance of rapamycin-loaded immunoliposomes in HER2-positive breast cancer models. Rapamycin-loaded immunoliposomes exhibit several distinct therapeutic advantages: 1) mTOR Pathway Inhibition: Rapamycin, a lipophilic macrolide antibiotic and potent mTOR inhibitor, targets a key regulatory pathway involved in mRNA translation, protein synthesis, glucose metabolism, and lipid synthesis. Dysregulation of the mTOR pathway is strongly associated with malignant progression in breast cancer. Trastuzumab-functionalized immunoliposomes enhance the delivery of rapamycin to HER2-overexpressing cells, intensifying its antitumour efficacy [124], [125], [126], [127]. 2) Improved Pharmacokinetics and Reduced Toxicity: Liposomal encapsulation reduces systemic toxicity, prolongs circulation time, and allows for lower therapeutic dosages by improving rapamycin’s pharmacokinetic profile. These features minimize pharmacological side effects and enhance patient compliance through optimized administration [128], [129], [130]. 3) Targeted Delivery via HER2 Receptor Specificity: Functionalization with trastuzumab enables the immunoliposomes to selectively bind to HER2 receptors, which are overexpressed in certain breast cancer subtypes. This receptor-specific targeting ensures selective drug delivery to tumour cells while sparing healthy tissues [131], [132], [133]. The development of rapamycin-loaded, trastuzumab-conjugated liposomal formulations necessitates the optimization of multiple physicochemical and biological parameters to ensure therapeutic efficacy. These include achieving nanometric particle size for effective tumour penetration, high drug loading efficiency, improved colloidal and storage stability, extended systemic circulation time. Conjugation of the antibody to the liposomal surface must be efficient and stable, without compromising the immunoreactivity of the antibody or the integrity of the liposome. Additional considerations include improved bioavailability, a favourable safety and toxicity profile, and demonstrably superior therapeutic efficacy compared to non-targeted or free drug formulations [134]. Rapamycin-loading studies began with the identification of the optimal drug-to-lipid ratio to attain nanometric particle sizes and elevate loading efficiency (Fig. 6). The results showed that using a 1:10 and 1:15 ratio of drug to SPC created small particles and high rapamycin encapsulation rates close to 90%. These results agree with Haeri et al, who found that using more lipid (DSPC) compared to rapamycin improved how well rapamycin was captured [135], [136]. SPC was found to be better at holding rapamycin, which is why this lipid was chosen to mix with Chol in different amounts. The formulations composed of SPC:Chol in the ratios of 10:2 and 10:3 exhibited an encapsulation efficiency of approximately 60% [137], [138]. It is important to remember that Chol also plays a critical role in maintaining liposomal stability [139]. The formulations made with 10:2 and 10:3 (SPC:Chol) probably had better stability because of Chol, which led to a higher amount of rapamycin being captured compared to the formulation made with 10:1 (SPC:Chol) [140].
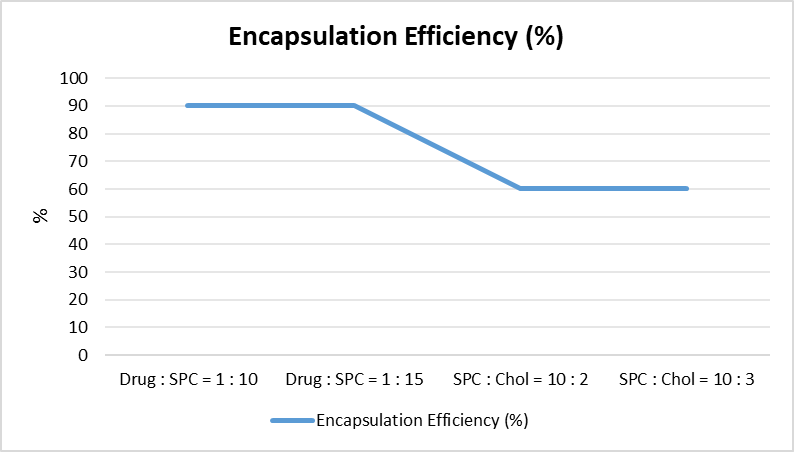
Fig. 6: Optimization of Rapamycin Encapsulation in SPC–Cholesterol Liposomal Formulations. Drug:SPC ratios of 1:10 and 1:15 produced nanometric particles with high encapsulation efficiency (~90%). Further modification with SPC:Chol ratios of 10:2 and 10:3 improved liposomal stability while maintaining encapsulation efficiency around ~60%.
DSPE-PEG 2000 can be added to the outside of liposomes to help them stay in the bloodstream longer, because polyethylene glycol (PEG), a stretchy water-loving material, reduces how much the mononuclear phagocytic system (MPS) takes up the nanoparticles. This results in an extended biological half-life and the accumulation of liposomes in solid tumours due to the enhanced permeability and retention (EPR) effect [141]. FTIR analysis was employed to characterize chemical bonding, identify functional groups, and evaluate intermolecular interactions within rapamycin-loaded liposomes [142]. Differential Scanning Calorimetry (DSC) was employed to investigate drug–carrier interactions, revealing the transformation of rapamycin from its crystalline to amorphous form within the nanocarrier system [141], [142]. The amorphous state is associated with increased solubility, potentially enhancing the drug's bioavailability. The colloidal stability of the PEGylated liposomes encapsulating rapamycin was assessed over a period of one month at 4 °C [143]. No significant changes were observed in particle size, polydispersity index, or encapsulation efficiency, indicating excellent formulation stability under these storage conditions [143], [144].
To enhance the specificity and therapeutic efficacy of liposomal drug delivery in cancer therapy, antibody-functionalized liposomes (immunoliposomes) were developed via covalent conjugation strategies. In this study, trastuzumab was thiolated using Traut’s reagent and successfully conjugated to DSPE-PEG-maleimide on the liposome surface, enabling stable and targeted immunoliposome formation (Fig. 7) [145], [146], [147], [148]. The structural integrity of trastuzumab following liposome functionalization was confirmed by SDS-PAGE under reducing conditions, demonstrating successful incorporation of intact antibody on the liposome surface. These results are consistent with previous findings by Yang et al. (2007), who reported similar outcomes in trastuzumab-functionalized paclitaxel-loaded immunoliposomes [149].

Fig. 7: Schematic representation of the immunoliposome reaction.
The cytotoxic effects of rapamycin, trastuzumab, and their respective formulations were evaluated in SK-BR-3, a HER2-overexpressing breast cancer cell line. SK-BR-3 cells exhibited high sensitivity to rapamycin, which can be attributed to HER2 amplification and subsequent activation of the mTOR signalling pathway [150]. Notably, rapamycin-loaded liposomes demonstrated cytotoxicity comparable to that of the free drug, highlighting the potential benefits of liposomal encapsulation, such as improved pharmacokinetics and reduced systemic toxicity [151], [152]. The combination of rapamycin and trastuzumab, administered either as free drugs or within immunoliposomes, significantly enhanced cytotoxicity in SK-BR-3 cells. This synergy was more pronounced in the immunoliposome formulation, as confirmed by the combination index (CI) determined using the Chou–Talalay method, yielding CI values of 0.15 for immunoliposomes and 0.29 for the free drug combination, indicating a strong synergistic interaction [153], [154], [155]. Although cytotoxicity in normal cells was not evaluated, reduced off-target effects are anticipated due to the lack of HER2 expression and lower proliferative rate in non-cancerous cells. The clinical relevance of combining mTOR inhibitors with trastuzumab has been supported by recent studies. A Phase II clinical trial demonstrated that daily administration of rapamycin with trastuzumab was well tolerated and suggested that mTOR inhibition may overcome resistance to trastuzumab in some HER2-positive tumours [155]. Additionally, combinations of everolimus—a rapamycin analog—with trastuzumab and paclitaxel have shown promising results in Phase I and II trials, and are currently under evaluation in the Phase III BOLERO-1 trial [156], [157]. In the research article by Arya et al. (2024), the therapeutic efficacy of targeted liposomes was investigated in HER2-positive SK-BR-3 human breast cancer cells using the drug trastuzumab. The cytotoxicity and drug delivery potential were found to be more in targeted liposomes than in non-targeted liposomes. It was also found that the dual ligand liposomes were more effective in cytotoxicity than the other liposomes. The IC50 value was found to be less in dual ligand liposomes (iRGD and trastuzumab) (4.34 ± 0.32 µg/mL) than in the other liposomes (trastuzumab-only liposomes) (4.98 ± 0.36 µg/mL). The hemolysis was found to be less than 5%, proving the biocompatibility of the liposomes [158]. The synergistic effects observed in vitro, coupled with promising clinical evidence supporting mTOR inhibition in trastuzumab-resistant tumours, highlight the translational potential of this strategy. Continued preclinical optimization and clinical evaluation of such multifunctional nanocarriers will be essential to realize their full therapeutic promise, potentially setting a new benchmark for personalized and precision-based breast cancer treatment.
Synergistic Targeting of HER2 and EGFR in Breast Cancer: Therapeutic Potential of Trastuzumab and Nimotuzumab Combination
Recent studies underscore the role of monoclonal antibodies such as trastuzumab and nimotuzumab in enhancing adjuvant therapy outcomes, warranting further investigation into their synergistic potential and long-term efficacy. Nimotuzumab, a humanized anti-EGFR monoclonal antibody (h-R3), has demonstrated synergistic anti-tumour effects when combined with trastuzumab in HER2-positive breast cancer models. HER2 and EGFR, both members of the receptor tyrosine kinase (RTK) family, are key therapeutic targets in cancer [158], [159], [160]. EGFR inhibition has been shown to enhance the efficacy of HER2-targeted therapies in vitro and in BT-474 xenograft models. Unlike other EGFR-targeting antibodies, nimotuzumab exhibits selective binding to cells with moderate-to-high EGFR expression, sparing normal tissues and reducing off-target toxicity [161], [162], [163], [164]. By blocking EGF binding to the extracellular domain of EGFR, it inhibits downstream signalling pathways. Nimotuzumab is approved in several countries for the treatment of glioma and head and neck cancers and is noted for its superior clinical tolerability compared to other EGFR inhibitors. Recent studies have found that HER2/HER3, nuclear erythroid related factor-2 (NRF2), and reactive oxygen species (ROS) work together to help cells grow and multiply in different types of cancer [165], [166], [167]. Additionally, research shows that molecules in the receptor tyrosine kinases (RTKs) pathway, like PI3K and ERK1/2, can directly activate NRF2 [168], [169]. Numerous facets of RTK signalling are modulated by ROS, the levels of which are directly influenced by NRF2 [168]. Khalil et al. showed that using HER2-targeted immunotherapy drugs effectively killed ovarian cancer cells by greatly reducing NRF2 activity [170]. Thus, the anti-tumour efficacy of this combination therapy involves the inhibition of the HER2/ERK1/2 signalling system, which includes the efficient suppression of pHER2 and pERK1/2. Nimotuzumab additionally enhanced trastuzumab-induced reactive oxygen species production and inhibited NRF2. Consequently, we hypothesized that the HER2/ERK1/2 pathway may interact with the NRF2 antioxidant response pathway in BT-474 breast cancer cells. This suggests that the combination of trastuzumab and nimotuzumab may decrease the cross-talk of signalling pathways and demonstrate improved anti-tumour effects [171]. Trastuzumab exhibits a long serum half-life of approximately 28.5 days, with a typical dosing schedule of once every 3 weeks in clinical settings [172]. Nimotuzumab has a shorter half-life, ranging from 7 to 14 days [173]. Both antibodies are primarily cleared via the reticuloendothelial system and target-mediated drug disposition. Importantly, co-administration of these agents does not significantly alter each other’s PK profiles, supporting their use in combination without dose adjustment [174]. The combination of trastuzumab and nimotuzumab demonstrated enhanced inhibitory effects on HER2-positive breast cancer cell lines BT-474 and SK-BR-3, with significantly greater activity at 10 mg/mL compared to either agent alone. BT-474 cells showed greater sensitivity to the combination treatment than SK-BR-3 [175]. To further assess therapeutic efficacy, we conducted in vivo studies using nude mice bearing BT-474 xenografts. The combination therapy significantly reduced tumour growth relative to monotherapies. These in vivo findings corroborate the in vitro data, underscoring the enhanced antitumour efficacy of trastuzumab plus nimotuzumab in HER2-overexpressing breast cancer models [175].
Targeted Photodynamic Therapy and Imaging of HER2-Positive Breast Cancer Using Trastuzumab–Chlorine 6 Conjugates
Targeted cancer therapies have progressed notably with the introduction of antibody–drug conjugates (ADCs), which facilitate the precise delivery of cytotoxic agents to malignant cells, thereby reducing systemic toxicity. Among these, photodynamic therapy (PDT) has emerged as a promising, minimally invasive modality that utilizes light-activated photosensitizers to generate reactive oxygen species (ROS) for tumour ablation [176], [177]. However, conventional PDT lacks specificity, often resulting in collateral damage to healthy tissues. To enhance the tumour selectivity of PDT, antibody–photosensitizer conjugates (APCs) have been developed, integrating the specificity of monoclonal antibodies with the phototoxic potential of photosensitizers [178]. Trastuzumab, a humanized monoclonal antibody targeting the HER2 receptor, is widely used in the treatment of HER2-positive breast cancer and has shown potential as a carrier for targeted delivery in APCs [179]. Chlorin e6 (Ce6), a second-generation photosensitizer, exhibits strong absorption in the near-infrared region, high singlet oxygen yield, and favourable pharmacokinetics, making it an attractive candidate for conjugation with antibodies. In a study conducted by Li et al. (2025), trastuzumab-based antibody photosensitizer conjugate proteins containing photosensitizers like SOPP3 and miniSOG, which are genetically encoded photosensitizers, were designed and engineered to specifically target and destroy HER2-positive breast cancer cells. The photochemical activity of the photosensitizer conjugate proteins was verified through the emission of singlet oxygen at 1270 nm when exposed to light. Cell viability in HER2-positive breast cancer cell models was significantly reduced by more than 50% when the conjugate proteins were exposed to light, whereas minimal effects were observed in non-irradiated and HER2-negative breast cancer cell models. Cell growth suppression was observed for more than 24 h [180]. In order to enhance the penetration and distribution of trastuzumab (Tra) within tumour tissues, A trastuzumab-chlorin e6 conjugate (TMPC) was created by utilizing a cross-linker consisting of maleimide and poly(ethylene glycol) [180]. This conjugate was designed to deliver Tra deeply into tumour tissues and promote Tra accumulation through the application of photodynamic therapy (PDT). Chlorin e6 (Ce6) in TMPC serves as a photosensitizer, exhibiting fluorescence at wavelengths of 660–670 nm, hence functioning as a fluorescence dye [180], [181], [182]. Ce6 functions as a tissue penetration enhancer, as the formation of reactive oxygen species (ROS) during laser irradiation destroys the extracellular matrix, facilitating deeper penetration of Ce6 [183]. Furthermore, the produced cytotoxic reactive oxygen species directly eliminate tumour cells by apoptosis or necrosis [176], [177]. Ce6 facilitates deeper penetration of Tra while simultaneously inducing tumour cell apoptosis; hence, TMPC enhances the tissue penetration capability and therapeutic efficiency of Tra in HER2-positive breast cancer [180]. In a study by Kim et al. (2018), a trastuzumab chlorin e6 conjugate, TMPC, was synthesized to improve the efficacy of targeted photodynamic therapy in a model of HER2-positive breast cancer. TMPC was found to accumulate sixfold more in SK-BR-3 xenograft mice at 24 hours compared to PEG-Ce6, showing a prolonged retention profile, thus showing better tumor localization of the trastuzumab conjugate. IIn vitro, TMPC was observed to have a specific binding activity to HER2 and was efficient in generating singlet oxygen upon irradiation at 660-670 nm, exhibiting a photo-reaction that was efficient enough to kill cancer cells through the generation of reactive oxygen species, resulting in apoptosis and necrosis of cancer cells [181]. Previous studies have reported the synthesis of poly(ethylene glycol)-chlorin e6 (PEG-Ce6) via conventional carbodiimide coupling [184], [185]. A 1:1 molar ratio of PEG to Ce6 was used to achieve the desired degree of substitution. The resulting PEG-Ce6 conjugate was purified using a Sephadex® LH-20 column. PEGylation significantly improved the water solubility of Ce6, suggesting the potential utility of this approach for antibody conjugation under aqueous conditions [186]. Similarly, 2-methacryloyloxyethyl phosphorylcholine (MPC) was synthesized via the same carbodiimide-mediated coupling strategy, and its structure was confirmed by ¹H NMR analysis [187]. To take use of both trastuzumab's selective binding and chlorin e6's photosensitizing capabilities, researchers created Tra-Ce6 conjugates [188]. The simple maleimide-thiol process was used to synthesize TMPC, and a molar ratio of 1-4:1 was determined for the MPC:Tra. The concentrations of Ce6 and Tra in TMPC were measured with a spectrophotometer and a BCA protein assay, with concentrations of 53.7 μg and 237.3 μg per mL, respectively. The diagnostic potential of TMPC was evaluated by measuring its fluorescence intensity. To validate the applicability of TMPC in photodynamic therapy (PDT) as a penetration agent, singlet oxygen generation was assessed under 670 nm laser irradiation using singlet oxygen sensor green (SOSG) [189], [190]. TMPC exhibited a 9-fold increase in singlet oxygen generation compared to MPC. In MDA-MB-231 cells, minimal fluorescence was detected for both MPC and TMPC regardless of laser exposure, indicating limited uptake or activation. Conversely, in SK-BR-3 cells, TMPC showed significant green fluorescence upon irradiation, indicating efficient ROS generation. This effect is attributed to the specific binding of the Tra component in TMPC to the HER2 receptor, which is overexpressed in SK-BR-3 cells [189], [190]. TMPC demonstrates potential as a diagnostic agent for breast adenocarcinoma through selective tissue detection based on HER2 receptor status. Due to the trastuzumab moiety incorporated in its structure, TMPC exhibits high binding affinity to HER2-overexpressing tumour cells, such as SK-BR-3 and BT-474 [191], [192]. This targeted interaction enables selective accumulation of TMPC in HER2-positive tissues, facilitating precise tumour localization. The associated Ce6 fluorophore further enhances this application by providing strong and specific fluorescence signals upon activation, allowing for real-time imaging and delineation of tumour margins [193]. Therefore, TMPC holds promise as a molecular imaging probe for the detection and diagnosis of HER2-positive breast adenocarcinoma, supporting its use in both preoperative tumour mapping and intraoperative guidance. At concentrations higher than 1 μg/mL, TMPC showed lethal effects in SK-BR-3 and BT-474 breast cancer cell lines that overexpress HER2. Laser irradiation was performed at intensities ranging from 0 to 10 J/cm² after a sub-cytotoxic dosage of 0.1 μg/mL was used for photodynamic evaluations. Treatment with TMPC at the maximum intensity of 10 J/cm² increased ROS production and cytotoxicity in both cell lines by a factor of five. Based on these findings, it appears that TMPC mainly causes cell death by ROS-mediated membrane damage when activated by light, and it specifically targets HER2-positive tumour cells [194]. Ce6 fluorescence was also found to be higher in SK-BR-3 tumours, suggesting that the tumours were able to penetrate and activate the tissue [195]. TMPC embodies a next-generation therapeutic paradigm that synergizes targeted delivery, photodynamic cytotoxicity, and diagnostic imaging in the treatment of HER2-positive breast cancer. Its dual therapeutic and diagnostic (theranostic) capabilities hold significant promise for improving clinical outcomes through enhanced tumour specificity, minimized off-target effects, and real-time visualization of tumour margins. Continued preclinical and clinical research will be crucial in advancing this promising modality toward routine oncological practice.
CONCLUSION
Trastuzumab has revolutionized the treatment landscape of HER2-positive breast cancer, significantly improving patient survival and redefining therapeutic strategies. However, its clinical efficacy is frequently limited by both intrinsic and acquired resistance mechanisms, necessitating the development of more robust and targeted combination approaches. This systematic review underscores the growing body of evidence supporting the use of trastuzumab in combination with agents such as pertuzumab, T-DM1, T-DXd, mTOR inhibitors, EGFR inhibitors like nimotuzumab, and innovative nanotechnologies including immunoliposomes and antibody–photosensitizer conjugates. These strategies enhance antitumour efficacy through complementary mechanisms—ranging from dual HER2 blockade and PI3K/Akt/mTOR pathway inhibition to targeted drug delivery and ROS-mediated cytotoxicity—while also improving pharmacokinetics and minimizing systemic toxicity. In particular, the integration of trastuzumab into antibody–drug conjugates and multifunctional nanocarriers offers promising avenues for overcoming resistance and expanding therapeutic options to HER2-low and trastuzumab-refractory populations. Continued preclinical optimization and rigorous clinical validation of these combination therapies are essential to translate these innovations into practice. Future research should focus on identifying predictive biomarkers, refining patient selection criteria, minimizing treatment-related adverse events, and personalizing therapy to achieve maximal clinical benefit. The evolving paradigm of trastuzumab-based combinations not only holds the potential to improve survival but also to transform HER2-positive breast cancer management into a more precise, durable, and patient-centered approach.
NOVELTY
The present study offers novelty by providing a comprehensive and integrative synthesis of emerging trastuzumab-based therapeutic strategies for HER2-positive breast cancer, emphasizing mechanistic synergy and advanced drug-delivery approaches. Unlike conventional reviews that primarily focus on the clinical efficacy of individual HER2-targeted agents, this work systematically examines the biological rationale and therapeutic implications of combining trastuzumab with multiple modalities, including dual HER2 blockade, antibody–drug conjugates, pathway-specific inhibitors, and nanotechnology-based delivery systems. A distinctive aspect of the study is the detailed integration of molecular signalling mechanisms—particularly those involving the PI3K/AKT/mTOR and HER2/ERK pathways—with therapeutic outcomes, thereby clarifying how these combinations may overcome intrinsic and acquired resistance to trastuzumab. Furthermore, the study highlights the translational potential of nanomedicine platforms such as trastuzumab-functionalized immunoliposomes and antibody–photosensitizer conjugates, which enable targeted drug delivery, improved pharmacokinetic performance, and the possibility of theranostic applications including imaging and photodynamic therapy. By combining evidence from preclinical models, mechanistic studies, and clinical trials, the work establishes a multidimensional perspective on HER2-targeted therapy rather than evaluating therapies in isolation. This integrative framework advances current knowledge by linking molecular oncology, targeted immunotherapy, and nanotechnology, thereby offering a clearer conceptual basis for the development of next-generation therapeutic strategies. Consequently, the study contributes novel insight into how trastuzumab-centered combination regimens can be optimized to enhance tumour selectivity, mitigate resistance mechanisms, and improve long-term clinical outcomes in patients with HER2-positive breast cancer.
REFERENCES




Shikhar Joshi*, Ekata Joshi, Visheshata Joshi, Naveen Sharma, Revisiting HER2 Targeting: Clinical and Translational Advances in Trastuzumab-Based Combination Therapy, Int. J. Med. Pharm. Sci., 2026, 2 (7), 814-847. https://doi.org/10.5281/zenodo.21398885
 10.5281/zenodo.21398885
10.5281/zenodo.21398885