
We use cookies to ensure our website works properly and to personalise your experience. Cookies policy
Department of Pharmacy Practice, Karpagam College of Pharmacy, Coimbatore-641032, Tamil Nadu, India
Plummer?Vinson syndrome (PVS), also known as Paterson?Brown?Kelly syndrome, is a rare clinical disorder characterized by the triad of post-cricoid dysphagia, iron-deficiency anemia, and upper esophageal webs. Although its exact etiology remains unclear, iron deficiency is considered central to its pathogenesis, leading to mucosal atrophy and web formation through disruption of iron-dependent enzymes and oxidative damage. Other proposed factors include nutritional deficiencies, autoimmune associations, and genetic predisposition. PVS primarily affects middle-aged women but may also occur in men and children. The condition has become less common with improved nutrition and iron fortification. This report presents a rare case of a 60-year-old male with progressive dysphagia for solids, oral ulcer, koilonychia, platynychia, and angular stomatitis. Laboratory findings revealed microcytic hypochromic anemia (Hb 8.4 g/dL), and endoscopy demonstrated a post-cricoid esophageal web at C5?C6, confirming PVS. The patient had a history of gastrojejunostomy, likely contributing to chronic iron malabsorption. Management included intravenous iron sucrose, blood transfusion, and vitamin supplementation, resulting in significant clinical improvement. PVS is clinically important due to its premalignant potential, with a 3?15% risk of developing squamous cell carcinoma of the hypopharynx or upper esophagus. Early recognition, correction of iron deficiency, and endoscopic dilation for persistent webs yield excellent outcomes. Long-term follow-up is essential for monitoring recurrence and malignancy risk. This case underscores the importance of considering PVS in patients presenting with dysphagia and iron-deficiency anemia, even in males and those with prior gastrointestinal surgeries.
Plummer–Vinson syndrome (PVS) is an uncommon clinical condition defined by the triad of post-cricoid dysphagia, iron deficiency anemia, and the presence of upper esophageal webs. [1][2][8][19] In the United Kingdom, it is known as Paterson–Brown–Kelly syndrome, named after British laryngologists Donald Ross Paterson (1863–1939) and Adam Brown–Kelly (1865–1941), who first reported the disorder in 1919. [1][2][9] Paterson reported the presence of esophageal webs and dysphagia and suggested the condition might be precancerous, while Brown–Kelly documented the characteristic triad. [8] The term Plummer–Vinson syndrome originates from two Mayo Clinic physicians, Henry Stanley Plummer (1874–1936) and Porter Paisley Vinson (1890–1959), who independently studied the disorder. In 1912, endocrinologist Plummer reported a condition termed “hysterical dysphagia,” characterized by upper esophageal spasms occurring in the absence of any structural obstruction. Later, in 1919, Vinson, a surgeon, attributed dysphagia to esophageal angulation and successfully treated it with dilation—hence the condition bears their name. [8] More than a hundred years later, the precise etiology of Plummer–Vinson syndrome is still not fully understood. Proposed etiological factors include nutritional deficiencies, particularly iron deficiency, along with genetic predisposition and autoimmune mechanisms. [1][3][7] Among these, iron deficiency has the strongest evidence, as numerous reports demonstrate improvements in dysphagia with iron therapy. It is thought that iron deficiency causes mucosal inflammation (mucositis), which contributes to the development of esophageal webs. Malnutrition and potential deficiencies in vitamin B have also been proposed as factors, though the evidence for these associations remains limited. The association of PVS with autoimmune diseases such as celiac disease, Crohn’s disease, rheumatoid arthritis, and thyroid disorders indicates a possible immune component in its pathogenesis, though the hypothesis remains unproven. In every patient, potential underlying causes of iron deficiency such as gastrointestinal bleeding or celiac disease should be carefully evaluated. [1][3] Because PVS is rare, epidemiological data regarding its incidence and prevalence are scarce, with most knowledge coming from individual case reports.[1] A significant population-based study conducted in South Wales during the 1960s reported the presence of post-cricoid webs in 0.3–1.1% of women and in 8.4–22.4% of women presenting with dysphagia, while no cases were identified in men.[1][3] Historically, PVS was more frequent in early 20th-century middle-aged Scandinavian women. The prevalence of the condition significantly decreased during the latter half of the 20th century, probably as a result of better nutritional standards and the widespread fortification of foods with iron. The disorder predominantly occurs in white populations, with women accounting for as much as 90% of cases in early Scandinavian research. It is most often diagnosed between the ages of 40 and 70, although rare cases have been reported in children. [1][8] The exact mechanism behind esophageal web formation in PVS is still not clearly understood. It is hypothesized that iron deficiency disrupts the function of iron-dependent enzymes, leading to oxidative stress, DNA damage, and myasthenic changes in the muscles involved in swallowing. These biochemical changes lead to mucosal atrophy and, over time, result in web formation as a consequence of recurrent epithelial damage. The web typically forms just below the cricopharyngeal muscle and is asymmetrically attached to the anterior wall of the esophagus. [1][8][19] Histopathologically, no inflammatory infiltrates are typically found, but fibrosis, epithelial atrophy, hyperplasia, hyperkeratosis, basal cell hyperplasia, and mild chronic inflammation are noted.[1] Iron deficiency has also been linked to reduced esophageal muscle contraction amplitude. Repeated mucosal trauma in the post-cricoid region during swallowing is believed to further contribute to web formation. Autoimmune mechanisms have been proposed as a contributing factor, given their association with disorders such as pernicious anemia, rheumatoid arthritis, celiac disease, and thyroiditis. The detection of thyroid autoantibodies in patients with PVS lends support to this theory, although further investigation is necessary. Additionally, some studies suggest that heterotopic gastric mucosa (inlet patches) might contribute to the condition, as ulceration and subsequent scarring in these areas could result in web-like strictures and iron depletion. However, histological studies rarely confirm gastric metaplasia. [1][3] Patients with PVS generally have an excellent prognosis. The majority of patients experience full relief of symptoms following a single session of esophagogastroduodenoscopy with dilatation, along with iron supplementation. However, PVS carries a significant risk estimated at 3–15% for developing squamous cell carcinoma of the hypopharynx or upper esophagus, likely due to chronic iron deficiency and resulting mucosal changes. [1][2][3][8][9] Diagnosis requires hematologic evaluation for iron deficiency anemia (CBC, peripheral smear, serum iron, ferritin, TIBC, and transferrin saturation).[1][2][3][8] Diagnostic and imaging options for PVS include barium swallow, video fluoroscopy which is considered the most accurate for identifying esophageal webs, fiberoptic endoscopy, and esophagoscopy [1]. Conditions that should be considered in the differential diagnosis include scleroderma, achalasia, diffuse esophageal spasm, pill-induced or inflammatory strictures, as well as chronic graft versus host disease and blistering skin disorders. [1][2][3] Management primarily aims to correct iron deficiency and alleviate dysphagia. In many cases, iron supplementation alone is sufficient to resolve symptoms, with endoscopic dilatation reserved for patients who continue to experience obstruction. Studies show high success rates, though recurrence may occur in 10–30% of cases. Resistant webs may require laser lysis. Usually, long-term symptom relief is achieved through dilation using bougies up to 17 mm or balloon dilatation, with the majority of patients requiring just one procedure. [1][2][8]
CASE SUMMARY:
A 60-year-old male patient presented with complaints of intermittent difficulty in swallowing for the past 3 months, which has progressively worsened over the last month, particularly with solid food intake. The condition is associated with a persistent mouth ulcer lasting for the past month, which is aggravated by intake of spicy foods. Increased appetite for the past month. He has a known history of systemic hypertension for the past 3 years but has not been adherent to his antihypertensive medications. He underwent a gastrojejunostomy 10 years ago. The patient also has a personal history of smoking and alcohol consumption for 10 years, which he quit 4 years ago. He has a past medical history of systemic hypertension for the past 3 years. On examination the patient was conscious and oriented. His vital signs were stable with a pulse rate of 87 beats per minute, a respiratory rate of 18 breaths per minute, and blood pressure of 120/80 mmHg. On systemic examination: CVS: S1, S2 heard, no murmur, RS: Bilateral normal vesicular breath sounds heard, no added sounds, P/A: Soft, non-tender, bowel sounds heard. Surgical scar measuring 10cm in length is seen extending from the epigastric the umbilicus umbilicus and CNS: Non-focal neurological deficit on further local examination: Spoon-shaped nails (koilonychia), flat nails (platynychia) and angular stomatitis.
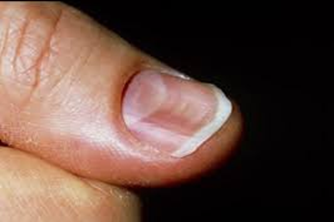
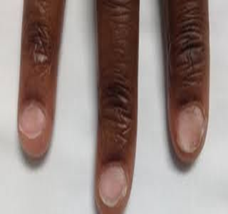
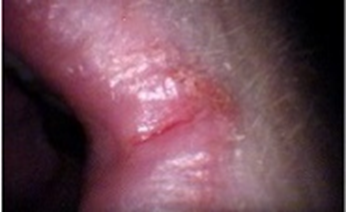
Figure 3. Angular stomatitis
On day of admission, the patient's initial investigations showed 8.4 g/dL in HB levels. Mean Corpuscular Volume (MCV): 65.2 fL, Mean Corpuscular Hemoglobin Concentration (MCHC): 27.0 g/dL, these findings are suggestive of microcytic hypochromic anemia, and an upper GI endoscopy showed a smooth, circular, whitish narrowing suggestive of a post-cricoid web at the C5 and C6 vertebral levels. The patient was diagnosed with iron deficiency anemia, Plummer-Vinson syndrome, and systemic hypertension. The patient was admitted for further evaluation and management. Anemia correction was initiated with Inj. Iron Sucrose 200 mg in 200 mL, Normal Saline STAT administered from day 2 to day 5. Four units of packed red blood cell (PRBC) transfusion are done on day 3. Pre-transfusion medications such as Inj. Avil 2cc and Inj. Dexa 4mg are administered (blood transfusion to the patient has been started at 3.15pm and completed by 5.35pm). The total iron deficit was found to be 1950 mg and it was corrected to 1600 mg; the remaining deficit is found to be 350 mg. T.Albendazole 400 mg, single dose (STAT); T. Folic Acid 5 mg once daily at 8 am from day 2 to day 5; T.Vitamin C 500 mg daily; T.Livogen XT once daily at 2 pm from day 1 to day 5; T.Supradyn multivitamin tablet once daily at 8 am from day 2 to day 5; T. Renerve Plus daily; T.Riboflavin daily. The patient's hemoglobin improved clinically, and he remained stable throughout hospitalization. The patient was discharged in a stable condition and prescribed iron supplements and nutritional supplements for 10 days. Advice on discharge: Consume plenty of fluids, follow a bland diet, and avoid spicy food.
DISCUSSION:
Plummer–Vinson syndrome (PVS) is an uncommon clinical entity characterized by a classic triad: post-cricoid dysphagia, iron-deficiency anemia, and the presence of upper esophageal webs. First recognized in the early 1900s, it is also referred to as Paterson–Brown–Kelly syndrome in the United Kingdom, named after the British clinicians who initially described it.[1][2][8] The condition primarily affects middle-aged women of Caucasian descent, typically between 40 and 70 years of age, with studies reporting a female predominance of up to 90%. However, PVS has occasionally been observed in males and even in pediatric populations. The present case of a 60-year-old male represents one of these rare instances, emphasizing that PVS should remain a diagnostic consideration regardless of sex or ethnicity. [2][5][8][1] Clinically, patients usually present with gradually progressive, painless dysphagia initially for solid foods, which may later extend to liquids over time. Dysphagia occurs when esophageal webs narrow the lumen to less than 12 mm. Additional features associated with iron deficiency, such as fatigue, pallor, koilonychia, glossitis, and angular stomatitis, are frequent and were present in this patient. Severe and chronic anemia may further produce systemic manifestations like tachycardia and generalized weakness. [8][1][2][16] Typically, the hallmark finding of an esophageal web is difficulty swallowing solids, with progression being gradual and predominantly affecting solid foods. Iron deficiency remains central to the disease pathogenesis. Since iron is vital for the activity of cellular enzymes involved in DNA synthesis and oxidative metabolism, its deficiency results in mucosal atrophy, epithelial degeneration, and ultimately the formation of post-cricoid esophageal webs. In this patient, a previous gastrojejunostomy may have contributed to chronic iron malabsorption, thereby exacerbating mucosal vulnerability. Additionally, reported associations between PVS and autoimmune diseases such as celiac disease, thyroiditis, and rheumatoid arthritis suggest an immunological component in disease development. [1][2][8][21] Following gastrojejunostomy, the altered gastrointestinal anatomy often leads to impaired iron absorption and progressive iron-deficiency anemia. Chronic iron depletion can induce atrophy of the esophageal mucosa, predisposing to the formation of thin, web-like membranes in the upper esophagus. In the current case, symptoms developed nearly a decade after the surgical procedure. [21] Histopathological examination of esophageal webs typically demonstrates epithelial atrophy, basal cell hyperplasia, fibrosis, and chronic inflammatory changes without significant inflammatory infiltrates, all consistent with mucosal injury secondary to iron deficiency. [1] Hematological findings in PVS generally reveal microcytic hypochromic anemia with reduced serum iron, ferritin, and transferrin saturation levels. Endoscopy serves as both a diagnostic and therapeutic tool, allowing visualization and potential dilation of esophageal webs, while video fluoroscopy can complement evaluation by assessing swallowing function. [2][8][1] In this patient, laboratory investigations revealed decreased MCV, MCHC, and hemoglobin levels, along with a microcytic hypochromic pattern on peripheral smear. On clinical examination, koilonychia (spoon nails), platynychia (flat nails), and angular stomatitis were noted. The patient’s history of one-month dysphagia corresponded with endoscopic findings of a smooth, circular, whitish narrowing in the post-cricoid region at the C5–C6 vertebral level, suggestive of an esophageal web. Iron supplementation remains the cornerstone of PVS management, frequently resulting in resolution of dysphagia and reversal of mucosal alterations. In this case, the patient showed significant improvement following intravenous iron therapy, blood transfusion, and vitamin supplementation. Endoscopic dilation is typically reserved for cases where obstruction persists despite correction of anemia. [8][1][2] A critical clinical consideration is that PVS is a precancerous condition, predisposing to squamous cell carcinoma of the hypopharynx and upper esophagus, with reported incidence ranging from 3% to 15%. Therefore, long-term surveillance endoscopy and modification of risk factors such as smoking and alcohol consumption are essential preventive measures. [1][2][8]
CONCLUSION:
Plummer-Vinson syndrome, also called Paterson-Kelly syndrome or sideropenic dysphagia, is a rare disorder linked to iron-deficiency anemia and the formation of thin, web-like membranes in the upper throat that can make swallowing difficult. Although the exact cause is unknown and prevention is uncertain, treating iron-deficiency anemia promptly and maintaining a diet rich in iron may help lower the risk. Most patients improve with iron therapy, which often resolves both anemia and swallowing problems. If swallowing remains difficult, a low-risk procedure called esophageal dilation can help, though some individuals may need multiple treatments for full relief. Outcomes are favourable with timely management; however, 10-30% recurrence rates exist, warranting long-term monitoring.
REFERENCES


Sakthi Thava Priya C.*, Lakshmanan M., Keerthana E., Roshini S., Logesh M., Anbuchelvan M., Neelesh P., A Rare Case Report on Plummer-Vinson Syndrome in A Post Gastrojejunostomy Geriatric Male Patient Int. J. Med. Pharm. Sci., 2025, 1 (11), 33-37. https://doi.org/10.5281/zenodo.17513476
 10.5281/zenodo.17513476
10.5281/zenodo.17513476